Carmona-Mora P et al. (OCT 2015)
Human Genetics 134 10 1099--1115
The nuclear localization pattern and interaction partners of GTF2IRD1 demonstrate a role in chromatin regulation
GTF2IRD1 is one of the three members of the GTF2I gene family,clustered on chromosome 7 within a 1.8 Mb region that is prone to duplications and deletions in humans. Hemizygous deletions cause Williams-Beuren syndrome (WBS) and duplications cause WBS duplication syndrome. These copy number variations disturb a variety of developmental systems and neurological functions. Human mapping data and analyses of knockout mice show that GTF2IRD1 and GTF2I underpin the craniofacial abnormalities,mental retardation,visuospatial deficits and hypersociability of WBS. However,the cellular role of the GTF2IRD1 protein is poorly understood due to its very low abundance and a paucity of reagents. Here,for the first time,we show that endogenous GTF2IRD1 has a punctate pattern in the nuclei of cultured human cell lines and neurons. To probe the functional relationships of GTF2IRD1 in an unbiased manner,yeast two-hybrid libraries were screened,isolating 38 novel interaction partners,which were validated in mammalian cell lines. These relationships illustrate GTF2IRD1 function,as the isolated partners are mostly involved in chromatin modification and transcriptional regulation,whilst others indicate an unexpected role in connection with the primary cilium. Mapping of the sites of protein interaction also indicates key features regarding the evolution of the GTF2IRD1 protein. These data provide a visual and molecular basis for GTF2IRD1 nuclear function that will lead to an understanding of its role in brain,behaviour and human disease.
View Publication
产品号#:
05850
05857
05870
05875
85850
85857
85870
85875
产品名:
mTeSR™1
mTeSR™1
Fuller HR et al. (JAN 2015)
Frontiers in cellular neuroscience 9 January 506
Spinal Muscular Atrophy Patient iPSC-Derived Motor Neurons Have Reduced Expression of Proteins Important in Neuronal Development.
Spinal muscular atrophy (SMA) is an inherited neuromuscular disease primarily characterized by degeneration of spinal motor neurons,and caused by reduced levels of the SMN protein. Previous studies to understand the proteomic consequences of reduced SMN have mostly utilized patient fibroblasts and animal models. We have derived human motor neurons from type I SMA and healthy controls by creating their induced pluripotent stem cells (iPSCs). Quantitative mass spectrometry of these cells revealed increased expression of 63 proteins in control motor neurons compared to respective fibroblasts,whereas 30 proteins were increased in SMA motor neurons vs. their fibroblasts. Notably,UBA1 was significantly decreased in SMA motor neurons,supporting evidence for ubiquitin pathway defects. Subcellular distribution of UBA1 was predominantly cytoplasmic in SMA motor neurons in contrast to nuclear in control motor neurons; suggestive of neurodevelopmental abnormalities. Many of the proteins that were decreased in SMA motor neurons,including beta III-tubulin and UCHL1,were associated with neurodevelopment and differentiation. These neuron-specific consequences of SMN depletion were not evident in fibroblasts,highlighting the importance of iPSC technology. The proteomic profiles identified here provide a useful resource to explore the molecular consequences of reduced SMN in motor neurons,and for the identification of novel biomarker and therapeutic targets for SMA.
View Publication
产品号#:
05832
05850
05857
05870
05875
85850
85857
85870
85875
产品名:
STEMdiff™ 神经花环选择试剂
mTeSR™1
mTeSR™1
Chamma I et al. (MAR 2016)
Nature Communications 7 10773
Mapping the dynamics and nanoscale organization of synaptic adhesion proteins using monomeric streptavidin
The advent of super-resolution imaging (SRI) has created a need for optimized labelling strategies. We present a new method relying on fluorophore-conjugated monomeric streptavidin (mSA) to label membrane proteins carrying a short,enzymatically biotinylated tag,compatible with SRI techniques including uPAINT,STED and dSTORM. We demonstrate efficient and specific labelling of target proteins in confined intercellular and organotypic tissues,with reduced steric hindrance and no crosslinking compared with multivalent probes. We use mSA to decipher the dynamics and nanoscale organization of the synaptic adhesion molecules neurexin-1β,neuroligin-1 (Nlg1) and leucine-rich-repeat transmembrane protein 2 (LRRTM2) in a dual-colour configuration with GFP nanobody,and show that these proteins are diffusionally trapped at synapses where they form apposed trans-synaptic adhesive structures. Furthermore,Nlg1 is dynamic,disperse and sensitive to synaptic stimulation,whereas LRRTM2 is organized in compact and stable nanodomains. Thus,mSA is a versatile tool to image membrane proteins at high resolution in complex live environments,providing novel information about the nano-organization of biological structures.
View Publication
产品号#:
05711
100-1281
产品名:
NeuroCult™ SM1 神经添加物
NeuroCult™ SM1 神经添加物
Kim YY et al. (SEP 2016)
PLOS ONE 11 9 e0163812
Alcohol-Induced Molecular Dysregulation in Human Embryonic Stem Cell-Derived Neural Precursor Cells
Adverse effect of alcohol on neural function has been well documented. Especially,the teratogenic effect of alcohol on neurodevelopment during embryogenesis has been demonstrated in various models,which could be a pathologic basis for fetal alcohol spectrum disorders (FASDs). While the developmental defects from alcohol abuse during gestation have been described,the specific mechanisms by which alcohol mediates these injuries have yet to be determined. Recent studies have shown that alcohol has significant effect on molecular and cellular regulatory mechanisms in embryonic stem cell (ESC) differentiation including genes involved in neural development. To test our hypothesis that alcohol induces molecular alterations during neural differentiation we have derived neural precursor cells from pluripotent human ESCs in the presence or absence of ethanol treatment. Genome-wide transcriptomic profiling identified molecular alterations induced by ethanol exposure during neural differentiation of hESCs into neural rosettes and neural precursor cell populations. The Database for Annotation,Visualization and Integrated Discovery (DAVID) functional analysis on significantly altered genes showed potential ethanol's effect on JAK-STAT signaling pathway,neuroactive ligand-receptor interaction,Toll-like receptor (TLR) signaling pathway,cytokine-cytokine receptor interaction and regulation of autophagy. We have further quantitatively verified ethanol-induced alterations of selected candidate genes. Among verified genes we further examined the expression of P2RX3,which is associated with nociception,a peripheral pain response. We found ethanol significantly reduced the level of P2RX3 in undifferentiated hESCs,but induced the level of P2RX3 mRNA and protein in hESC-derived NPCs. Our result suggests ethanol-induced dysregulation of P2RX3 along with alterations in molecules involved in neural activity such as neuroactive ligand-receptor interaction may be a molecular event associated with alcohol-related peripheral neuropathy of an enhanced nociceptive response.
View Publication
产品号#:
05850
05857
05870
05875
85850
85857
85870
85875
05835
05839
产品名:
mTeSR™1
mTeSR™1
STEMdiff™ 神经诱导培养基
STEMdiff™ 神经诱导培养基
Xu X et al. (MAR 2017)
Stem Cell Reports 8 3 619--633
Reversal of Phenotypic Abnormalities by CRISPR/Cas9-Mediated Gene Correction in Huntington Disease Patient-Derived Induced Pluripotent Stem Cells
Huntington disease (HD) is a dominant neurodegenerative disorder caused by a CAG repeat expansion in HTT. Here we report correction of HD human induced pluripotent stem cells (hiPSCs) using a CRISPR-Cas9 and piggyBac transposon-based approach. We show that both HD and corrected isogenic hiPSCs can be differentiated into excitable,synaptically active forebrain neurons. We further demonstrate that phenotypic abnormalities in HD hiPSC-derived neural cells,including impaired neural rosette formation,increased susceptibility to growth factor withdrawal,and deficits in mitochondrial respiration,are rescued in isogenic controls. Importantly,using genome-wide expression analysis,we show that a number of apparent gene expression differences detected between HD and non-related healthy control lines are absent between HD and corrected lines,suggesting that these differences are likely related to genetic background rather than HD-specific effects. Our study demonstrates correction of HD hiPSCs and associated phenotypic abnormalities,and the importance of isogenic controls for disease modeling using hiPSCs.
View Publication
CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons.
BACKGROUND Fragile X syndrome (FXS),a common cause of intellectual disability and autism,results from the expansion of a CGG-repeat tract in the 5' untranslated region of the FMR1 gene to<200 repeats. Such expanded alleles,known as full mutation (FM) alleles,are epigenetically silenced in differentiated cells thus resulting in the loss of FMRP,a protein important for learning and memory. The timing of repeat expansion and FMR1 gene silencing is controversial. METHODS We monitored the repeat size and methylation status of FMR1 alleles with expanded CGG repeats in patient-derived induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs) that were grown for extended period of time either as stem cells or differentiated into neurons. We used a PCR assay optimized for the amplification of large CGG repeats for sizing,and a quantitative methylation-specific PCR for the analysis of FMR1 promoter methylation. The FMR1 mRNA levels were analyzed by qRT-PCR. FMRP levels were determined by western blotting and immunofluorescence. Chromatin immunoprecipitation was used to study the association of repressive histone marks with the FMR1 gene in FXS ESCs. RESULTS We show here that while FMR1 gene silencing can be seen in FXS embryonic stem cells (ESCs),some silenced alleles contract and when the repeat number drops below ˜400,DNA methylation erodes,even when the repeat number remains<200. The resultant active alleles do not show the large step-wise expansions seen in stem cells from other repeat expansion diseases. Furthermore,there may be selection against large active alleles and these alleles do not expand further or become silenced on neuronal differentiation. CONCLUSIONS Our data support the hypotheses that (i) large expansions occur prezygotically or in the very early embryo,(ii) large unmethylated alleles may be deleterious in stem cells,(iii) methylation can occur on alleles with<400 repeats very early in embryogenesis,and (iv) expansion and contraction may occur by different mechanisms. Our data also suggest that the threshold for stable methylation of FM alleles may be higher than previously thought. A higher threshold might explain why some carriers of FM alleles escape methylation. It may also provide a simple explanation for why silencing has not been observed in mouse models with<200 repeats.
View Publication
产品号#:
05832
05850
05857
05870
05875
85850
85857
85870
85875
产品名:
STEMdiff™ 神经花环选择试剂
mTeSR™1
mTeSR™1
E. Gabriel et al. (JAN 2017)
Cell stem cell 20 3 397--406.e5
Recent Zika Virus Isolates Induce Premature Differentiation of Neural Progenitors in Human Brain Organoids.
The recent Zika virus (ZIKV) epidemic is associated with microcephaly in newborns. Although the connection between ZIKV and neurodevelopmental defects is widely recognized,the underlying mechanisms are poorly understood. Here we show that two recently isolated strains of ZIKV,an American strain from an infected fetal brain (FB-GWUH-2016) and a closely-related Asian strain (H/PF/2013),productively infect human iPSC-derived brain organoids. Both of these strains readily target to and replicate in proliferating ventricular zone (VZ) apical progenitors. The main phenotypic effect was premature differentiation of neural progenitors associated with centrosome perturbation,even during early stages of infection,leading to progenitor depletion,disruption of the VZ,impaired neurogenesis,and cortical thinning. The infection pattern and cellular outcome differ from those seen with the extensively passaged ZIKV strain MR766. The structural changes we see after infection with these more recently isolated viral strains closely resemble those seen in ZIKV-associated microcephaly.
View Publication
产品号#:
05832
05833
05790
05792
05793
05794
05795
85850
85857
85870
85875
05835
05839
08581
08582
产品名:
STEMdiff™ 神经花环选择试剂
STEMdiff™神经前体细胞培养基
BrainPhys™神经元培养基
BrainPhys™神经元培养基和SM1试剂盒
BrainPhys™ 神经元培养基N2-A和SM1试剂盒
BrainPhys™原代神经元试剂盒
BrainPhys™ hPSC 神经元试剂盒
mTeSR™1
mTeSR™1
STEMdiff™ 神经诱导培养基
STEMdiff™ 神经诱导培养基
STEMdiff™SMADi神经诱导试剂盒
STEMdiff™SMADi神经诱导试剂盒,2套
Chestkov IV et al. (JAN 2014)
Acta Naturae 6 1 54--60
The genetic reprogramming technology allows one to generate pluripotent stem cells for individual patients. These cells,called induced pluripotent stem cells (iPSCs),can be an unlimited source of specialized cell types for the body. Thus,autologous somatic cell replacement therapy becomes possible,as well as the generation of in vitro cell models for studying the mechanisms of disease pathogenesis and drug discovery. Amyotrophic lateral sclerosis (ALS) is an incurable neurodegenerative disorder that leads to a loss of upper and lower motor neurons. About 10% of cases are genetically inherited,and the most common familial form of ALS is associated with mutations in the SOD1 gene. We used the reprogramming technology to generate induced pluripotent stem cells with patients with familial ALS. Patient-specific iPS cells were obtained by both integration and transgene-free delivery methods of reprogramming transcription factors. These iPS cells have the properties of pluripotent cells and are capable of direct differentiation into motor neurons.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




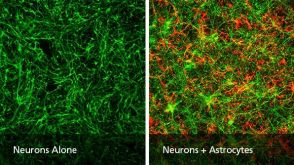

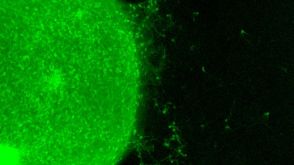 实验方案How to Generate AssemBloids™ from hPSC-Derived Dorsal and Ventral Forebrain Organoid Co-Cultures
实验方案How to Generate AssemBloids™ from hPSC-Derived Dorsal and Ventral Forebrain Organoid Co-Cultures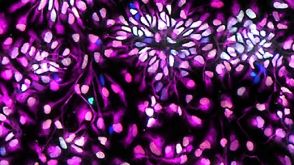

 沪公网安备31010102008431号
沪公网安备31010102008431号