
 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




产品号 #17899_C
免疫磁珠去除凋亡细胞(Annexin V)
若您需要咨询产品或有任何技术问题,请通过官方电话 400 885 9050 或邮箱 info.cn@stemcell.com 与我们联系。
免疫磁珠去除凋亡细胞(Annexin V)
免疫磁珠去除凋亡细胞(Annexin V)
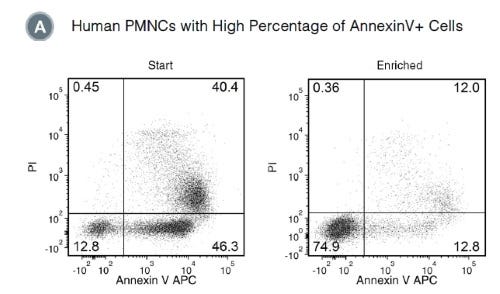
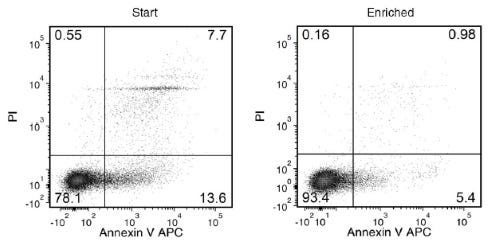
使用 EasySep™ 死细胞去除 (Annexin V) 试剂盒,通过免疫磁性负选,可高效去除细胞培养或组织制备样本中的凋亡细胞 (Annexin V+)。EasySep™结合了单克隆抗体的特异性和无柱磁性系统的简便性,迄今已广泛应用于发表的研究中超过20年。
EasySep™ 操作流程简单,使用识别 Annexin V的抗体复合物和磁珠标记细胞,再通过 EasySep™ 磁极进行无柱分选,只需倾倒或吸取未标记细胞,即可将标记细胞与未标记细胞分离,Annexin V+ 细胞则保留在管中。经过磁性细胞分离后,即可获得所需的细胞用于下游应用。在细胞凋亡过程中,Annexin V 会与细胞膜外层磷脂酰丝氨酸结合。
了解更多关于免疫磁性 EasySep™ 技术的工作原理。探索更多优化您实验流程的产品,包括培养基、补充剂、抗体等。
磁极兼容性
• EasySep™磁极(产品号 #18000),或
• “The Big Easy” EasySep™磁极(产品号 #18001),或
• EasyEights™ EasySep™磁极(产品号 #18103)
分类
细胞分选试剂盒
细胞类型
淋巴细胞
种属
人,小鼠,非人灵长类,其他组织,大鼠
样本来源
脐带血,白细胞单采术样本,肺,淋巴结,其他组织,脾脏
分选方法
去除
品牌
EasySep
研究领域
免疫
请在《产品说明书》中查找相关支持信息和使用说明,或浏览下方更多实验方案。
本产品专为以下研究领域设计,适用于工作流程中的高亮阶段。探索这些工作流程,了解更多我们为各研究领域提供的其他配套产品。
| 物种 | 人, 其它物种, 大鼠, 小鼠, 非人灵长类 |
|---|---|
| Magnet Compatibility | • EasySep™ Magnet (Catalog #18000), or • “The Big Easy” EasySep™ Magnet (Catalog #18001), or • EasyEights™ EasySep™ Magnet (Catalog #18103) |
| 样本来源 | 其它细胞系, 淋巴结, 白细胞单采术样本, 肺, 脐带血, 脾脏 |
| Selection Method | Depletion |
一种检测凋亡细胞的细胞蛋白

<p>通过免疫磁珠正选分离小鼠CD11b+细胞</p>

免疫磁珠负选不带标记的小鼠CD4+ T细胞

无菌聚苯乙烯细胞培养移液管

用于细胞分离和细胞培养的无菌聚苯乙烯圆底管;带锁扣帽或不带锁扣帽

用于细胞分离和细胞培养的无菌聚苯乙烯圆底管

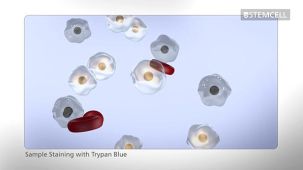
细胞活力染料(DNA标记染料)
在线联系






 沪公网安备31010102008431号
沪公网安备31010102008431号