Deonarain R et al. (NOV 2003)
Proceedings of the National Academy of Sciences of the United States of America 100 23 13453--8
Critical roles for IFN-beta in lymphoid development, myelopoiesis, and tumor development: links to tumor necrosis factor alpha.
We have generated mice null for IFN-beta and report the diverse consequences of IFN-beta for both the innate and adaptive arms of immunity. Despite no abnormalities in the proportional balance of CD4 and CD8 T cell populations in the peripheral blood,thymus,and spleen of IFN-beta-/- mice,activated lymph node and splenic T lymphocytes exhibit enhanced T cell proliferation and decreased tumor necrosis factor alpha production,relative to IFN-beta+/+ mice. Notably,constitutive and induced expression of tumor necrosis factor alpha is reduced in the spleen and bone marrow (BM) macrophages,respectively,of IFN-beta-/- mice. We also observe an altered splenic architecture in IFN-beta-/- mice and a reduction in resident macrophages. We identify a potential defect in B cell maturation in IFN-beta-/- mice,associated with a decrease in B220+ve/high/CD43-ve BM-derived cells and a reduction in BP-1,IgM,and CD23 expression. Circulating IgM-,Mac-1-,and Gr-1-positive cells are also substantially decreased in IFN-beta-/- mice. The decrease in the numbers of circulating macrophages and granulocytes likely reflects defective maturation of primitive BM hematopoiesis in mice,shown by the reduction of colony-forming units,granulocyte-macrophage. We proceeded to evaluate the in vivo growth of malignant cells in the IFN-beta-/- background and give evidence that Lewis lung carcinoma-specific tumor growth is more aggressive in IFN-beta-/- mice. Taken altogether,our data suggest that,in addition to the direct growth-inhibitory effects on tumor cells,IFN-beta is required during different stages of maturation in the development of the immune system.
View Publication
von Vietinghoff S et al. (MAY 2007)
Blood 109 10 4487--93
NB1 mediates surface expression of the ANCA antigen proteinase 3 on human neutrophils.
Antineutrophil cytoplasmic antibodies (ANCAs) with specificity for proteinase 3 (PR3) are central to a form of ANCA-associated vasculitis. Membrane PR3 (mPR3) is expressed only on a subset of neutrophils. The aim of this study was to determine the mechanism of PR3 surface expression on human neutrophils. Neutrophils were isolated from patients and healthy controls,and hematopoietic stem cells from cord blood served as a model of neutrophil differentiation. Surface expression was analyzed by flow cytometry and confocal microscopy,and proteins were analyzed by Western blot experiments. Neutrophil subsets were separated by magnetic cell sorting. Transfection experiments were carried out in HEK293 and HL60 cell lines. Using neutrophils from healthy donors,patients with vasculitis,and neutrophilic differentiated stem cells we found that mPR3 display was restricted to cells expressing neutrophil glycoprotein NB1,a glycosylphosphatidylinositol (GPI)-linked surface receptor. mPR3 expression was decreased by enzymatic removal of GPI anchors from cell membranes and was absent in a patient with paroxysmal nocturnal hemoglobinuria. PR3 and NB1 coimmunoprecipitated from and colocalized on the neutrophil plasma membrane. Transfection with NB1 resulted in specific PR3 surface binding in different cell types. We conclude that PR3 membrane expression on neutrophils is mediated by the NB1 receptor.
View Publication
产品号#:
09600
09650
产品名:
StemSpan™ SFEM
StemSpan™ SFEM
Bai M et al. ( 2017)
Blood 130 19 2092--2100
CD177 modulates human neutrophil migration through activation-mediated integrin and chemoreceptor regulation.
CD177 is a glycosylphosphatidylinositol (GPI)-anchored protein expressed by a variable proportion of human neutrophils that mediates surface expression of the antineutrophil cytoplasmic antibody antigen proteinase 3. CD177 associates with β2 integrins and recognizes platelet endothelial cell adhesion molecule 1 (PECAM-1),suggesting a role in neutrophil migration. However,CD177pos neutrophils exhibit no clear migratory advantage in vivo,despite interruption of in vitro transendothelial migration by CD177 ligation. We sought to understand this paradox. Using a PECAM-1-independent transwell system,we found that CD177pos and CD177neg neutrophils migrated comparably. CD177 ligation selectively impaired migration of CD177pos neutrophils,an effect mediated through immobilization and cellular spreading on the transwell membrane. Correspondingly,CD177 ligation enhanced its interaction with β2 integrins,as revealed by fluorescence lifetime imaging microscopy,leading to integrin-mediated phosphorylation of Src and extracellular signal-regulated kinase (ERK). CD177-driven cell activation enhanced surface β2 integrin expression and affinity,impaired internalization of integrin attachments,and resulted in ERK-mediated attenuation of chemokine signaling. We conclude that CD177 signals in a β2 integrin-dependent manner to orchestrate a set of activation-mediated mechanisms that impair human neutrophil migration.
View Publication
产品号#:
19666
100-0404
产品名:
EasySep™ Direct人中性粒细胞分选试剂盒
RoboSep™ 人中性粒细胞分选试剂盒
Hiyoshi H et al. (FEB 2018)
Cell reports 22 7 1787--1797
Mechanisms to Evade the Phagocyte Respiratory Burst Arose by Convergent Evolution in Typhoidal Salmonella Serovars.
Typhoid fever caused by Salmonella enterica serovar (S.) Typhi differs in its clinical presentation from gastroenteritis caused by S. Typhimurium and other non-typhoidal Salmonella serovars. The different clinical presentations are attributed in part to the virulence-associated capsular polysaccharide (Vi antigen) of S. Typhi,which prevents phagocytes from triggering a respiratory burst by preventing antibody-mediated complement activation. Paradoxically,the Vi antigen is absent from S. Paratyphi A,which causes a disease that is indistinguishable from typhoid fever. Here,we show that evasion of the phagocyte respiratory burst by S. Paratyphi A required very long O antigen chains containing the O2 antigen to inhibit antibody binding. We conclude that the ability to avoid the phagocyte respiratory burst is a property distinguishing typhoidal from non-typhoidal Salmonella serovars that was acquired by S. Typhi and S. Paratyphi A independently through convergent evolution.
View Publication
Novel imatinib-sensitive PDGFRA-activating point mutations in hypereosinophilic syndrome induce growth factor independence and leukemia-like disease.
The FIP1L1-PDGFRA fusion is seen in a fraction of cases with a presumptive diagnosis of hypereosinophilic syndrome (HES). However,because most HES patients lack FIP1L1-PDGFRA,we studied whether they harbor activating mutations of the PDGFRA gene. Sequencing of 87 FIP1L1-PDGFRA-negative HES patients revealed several novel PDGFRA point mutations (R481G,L507P,I562M,H570R,H650Q,N659S,L705P,R748G,and Y849S). When cloned into 32D cells,N659S and Y849S and-on selection for high expressors-also H650Q and R748G mutants induced growth factor-independent proliferation,clonogenic growth,and constitutive phosphorylation of PDGFRA and Stat5. Imatinib antagonized Stat5 phosphorylation. Mutations involving positions 659 and 849 had been shown previously to possess transforming potential in gastrointestinal stromal tumors. Because H650Q and R748G mutants possessed only weak transforming activity,we injected 32D cells harboring these mutants or FIP1L1-PDGFRA into mice and found that they induced a leukemia-like disease. Oral imatinib treatment significantly decreased leukemic growth in vivo and prolonged survival. In conclusion,our data provide evidence that imatinib-sensitive PDGFRA point mutations play an important role in the pathogenesis of HES and we propose that more research should be performed to further define the frequency and treatment response of PDGFRA mutations in FIP1L1-PDGFRA-negative HES patients.
View Publication
产品号#:
03231
产品名:
MethoCult™ M3231
Kim M-H et al. (MAR 2011)
Blood 117 12 3343--52
Neutrophil survival and c-kit(+)-progenitor proliferation in Staphylococcus aureus-infected skin wounds promote resolution.
Polymorphonuclear neutrophils (PMNs) are critical for the formation,maintenance,and resolution of bacterial abscesses. However,the mechanisms that regulate PMN survival and proliferation during the evolution of an abscess are not well defined. Using a mouse model of Staphylococcus aureus abscess formation within a cutaneous wound,combined with real-time imaging of genetically tagged PMNs,we observed that a high bacterial burden elicited a sustained mobilization of PMNs from the bone marrow to the infected wound,where their lifespan was markedly extended. A continuous rise in wound PMN number,which was not accounted for by trafficking from the bone marrow or by prolonged survival,was correlated with the homing of c-kit(+)-progenitor cells from the blood to the wound,where they proliferated and formed mature PMNs. Furthermore,by blocking their recruitment with an antibody to c-kit,which severely limited the proliferation of mature PMNs in the wound and shortened mouse survival,we confirmed that progenitor cells are not only important contributors to PMN expansion in the wound,but are also functionally important for immune protection. We conclude that the abscess environment provides a niche capable of regulating PMN survival and local proliferation of bone marrow-derived c-kit(+)-progenitor cells.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒








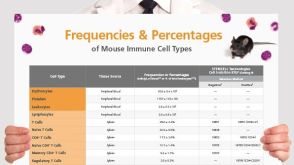 挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice
挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice

 沪公网安备31010102008431号
沪公网安备31010102008431号