Rawat VPS et al. (SEP 2010)
Proceedings of the National Academy of Sciences of the United States of America 107 39 16946--51
The vent-like homeobox gene VENTX promotes human myeloid differentiation and is highly expressed in acute myeloid leukemia.
Recent data indicate that a variety of regulatory molecules active in embryonic development may also play a role in the regulation of early hematopoiesis. Here we report that the human Vent-like homeobox gene VENTX,a putative homolog of the Xenopus xvent2 gene,is a unique regulatory hematopoietic gene that is aberrantly expressed in CD34(+) leukemic stem-cell candidates in human acute myeloid leukemia (AML). Quantitative RT-PCR documented expression of the gene in lineage positive hematopoietic subpopulations,with the highest expression in CD33(+) myeloid cells. Notably,expression levels of VENTX were negligible in normal CD34(+)/CD38(-) or CD34(+) human progenitor cells. In contrast to this,leukemic CD34(+)/CD38(-) cells from AML patients with translocation t(8,21) and normal karyotype displayed aberrantly high expression of VENTX. Gene expression and pathway analysis demonstrated that in normal CD34(+) cells enforced expression of VENTX initiates genes associated with myeloid development and down-regulates genes involved in early lymphoid development. Functional analyses confirmed that aberrant expression of VENTX in normal CD34(+) human progenitor cells perturbs normal hematopoietic development,promoting generation of myeloid cells and impairing generation of lymphoid cells in vitro and in vivo. Stable knockdown of VENTX expression inhibited the proliferation of human AML cell lines. Taken together,these data extend our insights into the function of embryonic mesodermal factors in human postnatal hematopoiesis and indicate a role for VENTX in normal and malignant myelopoiesis.
View Publication
产品号#:
04434
04444
产品名:
MethoCult™ H4434 Classic
MethoCult™ H4434 Classic
Fiedler K et al. (JAN 2011)
Blood 117 4 1329--39
Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia.
Bruton tyrosine kinase (Btk) is essential for B cell development and function and also appears to be important for myeloid cells. The bone marrow of Btk-deficient mice shows enhanced granulopoiesis compared with that of wild-type mice. In purified granulocyte-monocyte-progenitors (GMP) from Btk-deficient mice,the development of granulocytes is favored at the expense of monocytes. However,Btk-deficient neutrophils are impaired in maturation and function. Using bone marrow chimeras,we show that this defect is cell-intrinsic to neutrophils. In GMP and neutrophils,Btk plays a role in GM-CSF- and Toll-like receptor-induced differentiation. Molecular analyses revealed that expression of the lineage-determining transcription factors C/EBPα,C/EBPβ,and PU.1,depends on Btk. In addition,expression of several granule proteins,including myeloperoxidase,neutrophilic granule protein,gelatinase and neutrophil elastase,is Btk-dependent. In the Arthus reaction,an acute inflammatory response,neutrophil migration into tissues,edema formation,and hemorrhage are significantly reduced in Btk-deficient animals. Together,our findings implicate Btk as an important regulator of neutrophilic granulocyte maturation and function in vivo.
View Publication
Rara haploinsufficiency modestly influences the phenotype of acute promyelocytic leukemia in mice.
RARA (retinoic acid receptor alpha) haploinsufficiency is an invariable consequence of t(15;17)(q22;q21) translocations in acute promyelocytic leukemia (APL). Retinoids and RARA activity have been implicated in hematopoietic self-renewal and neutrophil maturation. We and others therefore predicted that RARA haploinsufficiency would contribute to APL pathogenesis. To test this hypothesis,we crossed Rara(+/-) mice with mice expressing PML (promyelocytic leukemia)-RARA from the cathepsin G locus (mCG-PR). We found that Rara haploinsufficiency cooperated with PML-RARA,but only modestly influenced the preleukemic and leukemic phenotype. Bone marrow from mCG-PR(+/-) × Rara(+/-) mice had decreased numbers of mature myeloid cells,increased ex vivo myeloid cell proliferation,and increased competitive advantage after transplantation. Rara haploinsufficiency did not alter mCG-PR-dependent leukemic latency or penetrance,but did influence the distribution of leukemic cells; leukemia in mCG-PR(+/-) × Rara(+/-) mice presented more commonly with low to normal white blood cell counts and with myeloid infiltration of lymph nodes. APL cells from these mice were responsive to all-trans retinoic acid and had virtually no differences in expression profiling compared with tumors arising in mCG-PR(+/-) × Rara(+/+) mice. These data show that Rara haploinsufficiency (like Pml haploinsufficiency and RARA-PML) can cooperate with PML-RARA to influence the pathogenesis of APL in mice,but that PML-RARA is the t(15;17) disease-initiating mutation.
View Publication
产品号#:
03534
产品名:
MethoCult™ GF M3534
Arai S et al. (JUN 2011)
Blood 117 23 6304--14
Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells.
Ecotropic viral integration site-1 (Evi-1) is a nuclear transcription factor that plays an essential role in the regulation of hematopoietic stem cells. Aberrant expression of Evi-1 has been reported in up to 10% of patients with acute myeloid leukemia and is a diagnostic marker that predicts a poor outcome. Although chromosomal rearrangement involving the Evi-1 gene is one of the major causes of Evi-1 activation,overexpression of Evi-1 is detected in a subgroup of acute myeloid leukemia patients without any chromosomal abnormalities,which indicates the presence of other mechanisms for Evi-1 activation. In this study,we found that Evi-1 is frequently up-regulated in bone marrow cells transformed by the mixed-lineage leukemia (MLL) chimeric genes MLL-ENL or MLL-AF9. Analysis of the Evi-1 gene promoter region revealed that MLL-ENL activates transcription of Evi-1. MLL-ENL-mediated up-regulation of Evi-1 occurs exclusively in the undifferentiated hematopoietic population,in which Evi-1 particularly contributes to the propagation of MLL-ENL-immortalized cells. Furthermore,gene-expression analysis of human acute myeloid leukemia cases demonstrated the stem cell-like gene-expression signature of MLL-rearranged leukemia with high levels of Evi-1. Our findings indicate that Evi-1 is one of the targets of MLL oncoproteins and is selectively activated in hematopoietic stem cell-derived MLL leukemic cells.
View Publication
产品号#:
03434
03444
产品名:
MethoCult™ GF M3434
MethoCult™ GF M3434
Nanua S et al. (MAR 2011)
Blood 117 13 3539--47
Activation of the unfolded protein response is associated with impaired granulopoiesis in transgenic mice expressing mutant Elane.
Severe congenital neutropenia (SCN) is an inborn disorder of granulopoiesis that in many cases is caused by mutations of the ELANE gene,which encodes neutrophil elastase (NE). Recent data suggest a model in which ELANE mutations result in NE protein misfolding,induction of endoplasmic reticulum (ER) stress,activation of the unfolded protein response (UPR),and ultimately a block in granulocytic differentiation. To test this model,we generated transgenic mice carrying a targeted mutation of Elane (G193X) reproducing a mutation found in SCN. The G193X Elane allele produces a truncated NE protein that is rapidly degraded. Granulocytic precursors from G193X Elane mice,though without significant basal UPR activation,are sensitive to chemical induction of ER stress. Basal and stress granulopoiesis after myeloablative therapy are normal in these mice. Moreover,inaction of protein kinase RNA-like ER kinase (Perk),one of the major sensors of ER stress,either alone or in combination with G193X Elane,had no effect on basal granulopoiesis. However,inhibition of the ER-associated degradation (ERAD) pathway using a proteosome inhibitor resulted in marked neutropenia in G193X Elane. The selective sensitivity of G913X Elane granulocytic cells to ER stress provides new and strong support for the UPR model of disease patho-genesis in SCN.
View Publication
产品号#:
03231
03434
03444
产品名:
MethoCult™ M3231
MethoCult™ GF M3434
MethoCult™ GF M3434
Mortensen M et al. (MAR 2011)
The Journal of experimental medicine 208 3 455--67
The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance.
The role of autophagy,a lysosomal degradation pathway which prevents cellular damage,in the maintenance of adult mouse hematopoietic stem cells (HSCs) remains unknown. Although normal HSCs sustain life-long hematopoiesis,malignant transformation of HSCs leads to leukemia. Therefore,mechanisms protecting HSCs from cellular damage are essential to prevent hematopoietic malignancies. In this study,we crippled autophagy in HSCs by conditionally deleting the essential autophagy gene Atg7 in the hematopoietic system. This resulted in the loss of normal HSC functions,a severe myeloproliferation,and death of the mice within weeks. The hematopoietic stem and progenitor cell compartment displayed an accumulation of mitochondria and reactive oxygen species,as well as increased proliferation and DNA damage. HSCs within the Lin(-)Sca-1(+)c-Kit(+) (LSK) compartment were significantly reduced. Although the overall LSK compartment was expanded,Atg7-deficient LSK cells failed to reconstitute the hematopoietic system of lethally irradiated mice. Consistent with loss of HSC functions,the production of both lymphoid and myeloid progenitors was impaired in the absence of Atg7. Collectively,these data show that Atg7 is an essential regulator of adult HSC maintenance.
View Publication
产品号#:
03434
03444
产品名:
MethoCult™ GF M3434
MethoCult™ GF M3434
Dixon AS et al. (AUG 2011)
The Journal of biological chemistry 286 31 27751--60
Disruption of Bcr-Abl coiled coil oligomerization by design.
Oligomerization is an important regulatory mechanism for many proteins,including oncoproteins and other pathogenic proteins. The oncoprotein Bcr-Abl relies on oligomerization via its coiled coil domain for its kinase activity,suggesting that a designed coiled coil domain with enhanced binding to Bcr-Abl and reduced self-oligomerization would be therapeutically useful. Key mutations in the coiled coil domain of Bcr-Abl were identified that reduce homo-oligomerization through intermolecular charge-charge repulsion yet increase interaction with the Bcr-Abl coiled coil through additional salt bridges,resulting in an enhanced ability to disrupt the oligomeric state of Bcr-Abl. The mutations were modeled computationally to optimize the design. Assays performed in vitro confirmed the validity and functionality of the optimal mutations,which were found to exhibit reduced homo-oligomerization and increased binding to the Bcr-Abl coiled coil domain. Introduction of the mutant coiled coil into K562 cells resulted in decreased phosphorylation of Bcr-Abl,reduced cell proliferation,and increased caspase-3/7 activity and DNA segmentation. Importantly,the mutant coiled coil domain was more efficacious than the wild type in all experiments performed. The improved inhibition of Bcr-Abl through oligomeric disruption resulting from this modified coiled coil domain represents a viable alternative to small molecule inhibitors for therapeutic intervention.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒





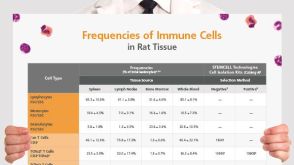 挂图Frequencies of Immune Cells in Rat Tissue Lists the estimated frequencies of more than 15 immune cell types in Sprague Dawley rats
挂图Frequencies of Immune Cells in Rat Tissue Lists the estimated frequencies of more than 15 immune cell types in Sprague Dawley rats 科学海报Isolation of Untouched Neutrophils Directly from Whole Blood in 20 Minutes
科学海报Isolation of Untouched Neutrophils Directly from Whole Blood in 20 Minutes

 沪公网安备31010102008431号
沪公网安备31010102008431号