Miething C et al. (MAR 2007)
Proceedings of the National Academy of Sciences of the United States of America 104 11 4594--9
Retroviral insertional mutagenesis identifies RUNX genes involved in chronic myeloid leukemia disease persistence under imatinib treatment.
The kinase inhibitor imatinib mesylate targeting the oncoprotein Bcr-Abl has revolutionized the treatment of chronic myeloid leukemia (CML). However,even though imatinib successfully controls the leukemia in chronic phase,it seems not to be able to cure the disease,potentially necessitating lifelong treatment with the inhibitor under constant risk of relapse. On a molecular level,the cause of disease persistence is not well understood. Initial studies implied that innate features of primitive progenitor cancer stem cells may be responsible for the phenomenon. Here,we describe an assay using retroviral insertional mutagenesis (RIM) to identify genes contributing to disease persistence in vivo. We transplanted mice with bone marrow cells retrovirally infected with the Bcr-Abl oncogene and subsequently treated the animals with imatinib to select for leukemic cells in which the proviral integration had affected genes modulating the imatinib response. Southern blot analysis demonstrated clonal outgrowth of cells carrying similar integration sites. Candidate genes located near the proviral insertion sites were identified,among them the transcription factor RUNX3. Proviral integration near the RUNX3 promoter induced RUNX3 expression,and Bcr-Abl-positive cell lines with stable or inducible expression of RUNX1 or RUNX3 were protected from imatinib-induced apoptosis. Furthermore,imatinib treatment selected for RUNX1-expressing cells in vitro and in vivo after infection of primary bone marrow cells with Bcr-Abl and RUNX1. Our results demonstrate the utility of RIM for probing molecular modulators of targeted therapies and suggest a role for members of the RUNX transcription factor family in disease persistence in CML patients.
View Publication
产品号#:
04230
产品名:
MethoCult™ H4230
Yang J et al. (SEP 2007)
Blood 110 6 2034--40
AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo.
Aurora kinases play an important role in chromosome alignment,segregation,and cytokinesis during mitosis. We have recently shown that hematopoietic malignant cells including those from acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) aberrantly expressed Aurora A and B kinases,and ZM447439,a potent inhibitor of Aurora kinases,effectively induced growth arrest and apoptosis of a variety of leukemia cells. The present study explored the effect of AZD1152,a highly selective inhibitor of Aurora B kinase,on various types of human leukemia cells. AZD1152 inhibited the proliferation of AML lines (HL-60,NB4,MOLM13),ALL line (PALL-2),biphenotypic leukemia (MV4-11),acute eosinophilic leukemia (EOL-1),and the blast crisis of chronic myeloid leukemia K562 cells with an IC50 ranging from 3 nM to 40 nM,as measured by thymidine uptake on day 2 of culture. These cells had 4N/8N DNA content followed by apoptosis,as measured by cell-cycle analysis and annexin V staining,respectively. Of note,AZD1152 synergistically enhanced the antiproliferative activity of vincristine,a tubulin depolymerizing agent,and daunorubicin,a topoisomerase II inhibitor,against the MOLM13 and PALL-2 cells in vitro. Furthermore,AZD1152 potentiated the action of vincristine and daunorubicin in a MOLM13 murine xenograft model. Taken together,AZD1152 is a promising new agent for treatment of individuals with leukemia. The combined administration of AZD1152 and conventional chemotherapeutic agent to patients with leukemia warrants further investigation.
View Publication
产品号#:
04564
04534
04544
产品名:
MethoCult™ H4534 Classic 无 EPO 入门试剂盒
MethoCult™ H4534 Classic(不含 EPO)
MethoCult™ H4534 Classic(不含 EPO)
Moulding DA et al. (SEP 2007)
The Journal of experimental medicine 204 9 2213--24
Unregulated actin polymerization by WASp causes defects of mitosis and cytokinesis in X-linked neutropenia.
Specific mutations in the human gene encoding the Wiskott-Aldrich syndrome protein (WASp) that compromise normal auto-inhibition of WASp result in unregulated activation of the actin-related protein 2/3 complex and increased actin polymerizing activity. These activating mutations are associated with an X-linked form of neutropenia with an intrinsic failure of myelopoiesis and an increase in the incidence of cytogenetic abnormalities. To study the underlying mechanisms,active mutant WASp(I294T) was expressed by gene transfer. This caused enhanced and delocalized actin polymerization throughout the cell,decreased proliferation,and increased apoptosis. Cells became binucleated,suggesting a failure of cytokinesis,and micronuclei were formed,indicative of genomic instability. Live cell imaging demonstrated a delay in mitosis from prometaphase to anaphase and confirmed that multinucleation was a result of aborted cytokinesis. During mitosis,filamentous actin was abnormally localized around the spindle and chromosomes throughout their alignment and separation,and it accumulated within the cleavage furrow around the spindle midzone. These findings reveal a novel mechanism for inhibition of myelopoiesis through defective mitosis and cytokinesis due to hyperactivation and mislocalization of actin polymerization.
View Publication
产品号#:
04100
产品名:
MethoCult™ H4100
Lidonnici MR et al. (MAY 2008)
Blood 111 9 4771--9
Requirement of c-Myb for p210(BCR/ABL)-dependent transformation of hematopoietic progenitors and leukemogenesis.
The c-Myb gene encodes a transcription factor required for proliferation and survival of normal myeloid progenitors and leukemic blast cells. Targeting of c-Myb by antisense oligodeoxynucleotides has suggested that myeloid leukemia blasts (including chronic myelogenous leukemia [CML]-blast crisis cells) rely on c-Myb expression more than normal progenitors,but a genetic approach to assess the requirement of c-Myb by p210(BCR/ABL)-transformed hematopoietic progenitors has not been taken. We show here that loss of a c-Myb allele had modest effects (20%-28% decrease) on colony formation of nontransduced progenitors,while the effect on p210(BCR/ABL)-expressing Lin(-) Sca-1(+) and Lin(-) Sca-1(+)Kit(+) cells was more pronounced (50%-80% decrease). Using a model of CML-blast crisis,mice (n = 14) injected with p210(BCR/ABL)-transduced p53(-/-)c-Myb(w/w) marrow cells developed leukemia rapidly and had a median survival of 26 days,while only 67% of mice (n = 12) injected with p210(BCR/ABL)-transduced p53(-/-)c-Myb(w/d) marrow cells died of leukemia with a median survival of 96 days. p210(BCR/ABL)-transduced c-Myb(w/w) and c-Myb(w/d) marrow progenitors expressed similar levels of the c-Myb-regulated genes c-Myc and cyclin B1,while those of Bcl-2 were reduced. However,ectopic Bcl-2 expression did not enhance colony formation of p210(BCR/ABL)-transduced c-Myb(w/d) Lin(-)Sca-1(+)Kit(+) cells. Together,these studies support the requirement of c-Myb for p210(BCR/ABL)-dependent leukemogenesis.
View Publication
产品号#:
04230
产品名:
MethoCult™ H4230
Z. Yan et al. (apr 2019)
JCI insight 5
Deficiency of Socs3 leads to brain-targeted EAE via enhanced neutrophil activation and ROS production.
Dysregulation of the JAK/STAT signaling pathway is associated with Multiple Sclerosis (MS) and its mouse model,Experimental Autoimmune Encephalomyelitis (EAE). Suppressors Of Cytokine Signaling (SOCS) negatively regulate the JAK/STAT pathway. We previously reported a severe,brain-targeted,atypical form of EAE in mice lacking Socs3 in myeloid cells (Socs3DeltaLysM),which is associated with cerebellar neutrophil infiltration. There is emerging evidence that neutrophils are detrimental in the pathology of MS/EAE,however,their exact function is unclear. Here we demonstrate that neutrophils from the cerebellum of Socs3DeltaLysM mice show a hyper-activated phenotype with excessive production of reactive oxygen species (ROS) at the peak of EAE. Neutralization of ROS in vivo delayed the onset and reduced severity of atypical EAE. Mechanistically,Socs3-deficient neutrophils exhibit enhanced STAT3 activation,a hyper-activated phenotype in response to G-CSF,and upon G-CSF priming,increased ROS production. Neutralization of G-CSF in vivo significantly reduced the incidence and severity of the atypical EAE phenotype. Overall,our work elucidates that hypersensitivity of G-CSF/STAT3 signaling in Socs3DeltaLysM mice leads to atypical EAE by enhanced neutrophil activation and increased oxidative stress,which may explain the detrimental role of G-CSF in MS patients.
View Publication
产品号#:
19762
19762RF
产品名:
EasySep™小鼠中性粒细胞富集试剂盒
RoboSep™ 小鼠中性粒细胞富集试剂盒含滤芯吸头
A. Gorgens et al. (May 2013)
Cell Reports 3 1539-1552
Revision of the Human Hematopoietic Tree: Granulocyte Subtypes Derive from Distinct Hematopoietic Lineages
The classical model of hematopoiesis predicts a dichotomous lineage restriction of multipotent hematopoietic progenitors (MPPs) into common lymphoid progenitors (CLPs) and common myeloid progenitors (CMPs). However,this idea has been challenged by the identification of lymphoid progenitors retaining partial myeloid potential (e.g.,LMPPs),implying that granulocytes can arise within both the classical lymphoid and the myeloid branches. Here,we resolve this issue by using cell-surface CD133 expression to discriminate functional progenitor populations. We show that eosinophilic and basophilic granulocytes as well as erythrocytes and megakaryocytes derive from a common erythro-myeloid progenitor (EMP),whereas neutrophilic granulocytes arise independently within a lympho-myeloid branch with long-term progenitor function. These findings challenge the concept of a CMP and restore dichotomy to the classical hematopoietic model.
View Publication
Clendening JW et al. (JUN 2010)
Blood 115 23 4787--97
Exploiting the mevalonate pathway to distinguish statin-sensitive multiple myeloma.
Statin inhibitors,used to control hypercholesterolemia,trigger apoptosis of hematologic tumor cells and therefore have immediate potential as anticancer agents. Evaluations of statins in acute myelogenous leukemia and multiple myeloma have shown that statin efficacy is mixed,with only a subset of tumor cells being highly responsive. Our goal was to distinguish molecular features of statin-sensitive and -insensitive myeloma cells and gain insight into potential predictive markers. We show that dysregulation of the mevalonate pathway is a key determinant of sensitivity to statin-induced apoptosis in multiple myeloma. In sensitive cells,the classic feedback response to statin exposure is lost. This results in deficient up-regulation of 2 isoforms of hydroxymethylglutaryl coenzyme A reductase: the rate-limiting enzyme of the mevalonate pathway and hydroxymethylglutaryl coenzyme A synthase 1. To ascertain the clinical utility of these findings,we demonstrate that a subset of primary myeloma cells is sensitive to statins and that monitoring dysregulation of the mevalonate pathway may distinguish these cancers. We also show statins are highly effective and well tolerated in an orthotopic model of myeloma using cells harboring this dysregulation. This determinant of sensitivity further provides molecular rationale for the significant therapeutic index of statins on these tumor cells.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒





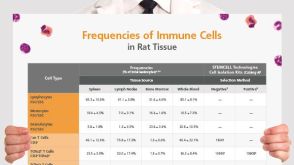 挂图Frequencies of Immune Cells in Rat Tissue Lists the estimated frequencies of more than 15 immune cell types in Sprague Dawley rats
挂图Frequencies of Immune Cells in Rat Tissue Lists the estimated frequencies of more than 15 immune cell types in Sprague Dawley rats


 沪公网安备31010102008431号
沪公网安备31010102008431号