Fujii T et al. (SEP 2010)
The Journal of biological chemistry 285 39 30214--23
Essential role of p400/mDomino chromatin-remodeling ATPase in bone marrow hematopoiesis and cell-cycle progression.
p400/mDomino is an ATP-dependent chromatin-remodeling protein that catalyzes the deposition of histone variant H2A.Z into nucleosomes to regulate gene expression. We previously showed that p400/mDomino is essential for embryonic development and primitive hematopoiesis. Here we generated a conditional knock-out mouse for the p400/mDomino gene and investigated the role of p400/mDomino in adult bone marrow hematopoiesis and in the cell-cycle progression of embryonic fibroblasts. The Mx1-Cre- mediated deletion of p400/mDomino resulted in an acute loss of nucleated cells in the bone marrow,including committed myeloid and erythroid cells as well as hematopoietic progenitor and stem cells. A hematopoietic colony assay revealed a drastic reduction in colony-forming activity after the deletion of p400/mDomino. Moreover,the loss of p400/mDomino in mouse embryonic fibroblasts (MEFs) resulted in strong growth inhibition. Cell-cycle analysis revealed that the mDomino-deficient MEFs exhibited a pleiotropic cell-cycle defect at the S and G(2)/M phases,and polyploid and multi-nucleated cells with micronuclei emerged. DNA microarray analysis revealed that the p400/mDomino deletion from MEFs caused the impaired expression of many cell-cycle-regulatory genes,including G(2)/M-specific genes targeted by the transcription factors FoxM1 and c-Myc. These results indicate that p400/mDomino plays a key role in cellular proliferation by controlling the expression of cell-cycle-regulatory genes.
View Publication
产品号#:
03434
03444
产品名:
MethoCult™ GF M3434
MethoCult™ GF M3434
Chin JY et al. (SEP 2008)
Proceedings of the National Academy of Sciences of the United States of America 105 36 13514--9
Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids.
Splice-site mutations in the beta-globin gene can lead to aberrant transcripts and decreased functional beta-globin,causing beta-thalassemia. Triplex-forming DNA oligonucleotides (TFOs) and peptide nucleic acids (PNAs) have been shown to stimulate recombination in reporter gene loci in mammalian cells via site-specific binding and creation of altered helical structures that provoke DNA repair. We have designed a series of triplex-forming PNAs that can specifically bind to sequences in the human beta-globin gene. We demonstrate here that these PNAs,when cotransfected with recombinatory donor DNA fragments,can promote single base-pair modification at the start of the second intron of the beta-globin gene,the site of a common thalassemia-associated mutation. This single base pair change was detected by the restoration of proper splicing of transcripts produced from a green fluorescent protein-beta-globin fusion gene. The ability of these PNAs to induce recombination was dependent on dose,sequence,cell-cycle stage,and the presence of a homologous donor DNA molecule. Enhanced recombination,with frequencies up to 0.4%,was observed with use of the lysomotropic agent chloroquine. Finally,we demonstrate that these PNAs were effective in stimulating the modification of the endogenous beta-globin locus in human cells,including primary hematopoietic progenitor cells. This work suggests that PNAs can be effective tools to induce heritable,site-specific modification of disease-related genes in human cells.
View Publication
产品号#:
02690
09600
09650
产品名:
StemSpan™ CC100
StemSpan™ SFEM
StemSpan™ SFEM
Cohen-Haguenauer O et al. (FEB 2006)
Proceedings of the National Academy of Sciences of the United States of America 103 7 2340--5
In vivo repopulation ability of genetically corrected bone marrow cells from Fanconi anemia patients.
Fanconi anemia (FA) is a rare inherited genomic instability syndrome representing one of the best examples of hematopoietic stem cell deficiency. Although FA might be an excellent candidate for bone marrow (BM) genetic correction ex vivo,knockout animal models are not sufficient to guide preclinical steps,and gene therapy attempts have proven disappointing so far. Contributing to these poor results is a characteristic and dramatic early BM-cells die-off when placed in culture. We show here that human primary FA BM cell survival can be ameliorated by using specific culture conditions that limit oxidative stress. When coupled with retrovirus-mediated transfer of the main complementation group FANCA-cDNA,we could achieve long-term reconstitution of the stem cell compartment both in vitro and in vivo. Gene-corrected BM cultures grew for textgreater120 days,and after cultured cell transplantation into NOD/SCID mice,clonogenic human cells carrying the FANCA transgene could be detected 6 months after transduction. By comparison,untransduced cells died in culture by 15 days. Of necessity for ethical reasons,experiments were conducted on a very limited number of primary BM cells. By using low cytokine regimen and conditions matching regulatory requirements,a contingent of gene-corrected cells slowly emerges with an unmet potential for in vivo engraftment. Future therapeutic applications of stem cells might be expanding from these data. In addition,we provide a model of gene-corrected human primary cell growth that carries the potential to better delineate the combined role of both DNA damage and oxidative stress in the pathogenesis of FA.
View Publication
产品号#:
04436
产品名:
MethoCult™ SF H4436
Capoccia BJ et al. (MAY 2009)
Blood 113 21 5340--51
Revascularization of ischemic limbs after transplantation of human bone marrow cells with high aldehyde dehydrogenase activity.
The development of cell therapies to treat peripheral vascular disease has proven difficult because of the contribution of multiple cell types that coordinate revascularization. We characterized the vascular regenerative potential of transplanted human bone marrow (BM) cells purified by high aldehyde dehydrogenase (ALDH(hi)) activity,a progenitor cell function conserved between several lineages. BM ALDH(hi) cells were enriched for myelo-erythroid progenitors that produced multipotent hematopoietic reconstitution after transplantation and contained nonhematopoietic precursors that established colonies in mesenchymal-stromal and endothelial culture conditions. The regenerative capacity of human ALDH(hi) cells was assessed by intravenous transplantation into immune-deficient mice with limb ischemia induced by femoral artery ligation/transection. Compared with recipients injected with unpurified nucleated cells containing the equivalent of 2- to 4-fold more ALDH(hi) cells,mice transplanted with purified ALDH(hi) cells showed augmented recovery of perfusion and increased blood vessel density in ischemic limbs. ALDH(hi) cells transiently recruited to ischemic regions but did not significantly integrate into ischemic tissue,suggesting that transient ALDH(hi) cell engraftment stimulated endogenous revascularization. Thus,human BM ALDH(hi) cells represent a progenitor-enriched population of several cell lineages that improves perfusion in ischemic limbs after transplantation. These clinically relevant cells may prove useful in the treatment of critical ischemia in humans.
View Publication
产品号#:
01700
01705
01701
01702
18058
18058RF
21000
20119
20155
产品名:
ALDEFLUOR™ 试剂盒
ALDEFLUOR™ DEAB试剂, 1.5 mM, 1 mL
ALDEFLUOR™检测缓冲液
RoboSep™- S
RoboSep™ 吸头组件抛光剂
RoboSep™分选管套装(9个塑料管)
Rutella S et al. (JUL 2006)
Blood 108 1 218--27
Hepatocyte growth factor favors monocyte differentiation into regulatory interleukin (IL)-10++IL-12low/neg accessory cells with dendritic-cell features.
Several hematopoietic growth factors,including interleukin-10 (IL-10) and transforming growth factor-beta1 (TGF-beta1),promote the differentiation of tolerogenic dendritic cells (DCs). Hepatocyte growth factor (HGF) is a pleiotropic cytokine whose effects on human DC differentiation and function have not been investigated. Monocytes cultured with HGF (HGFMo) differentiated into accessory cells with DC-like morphology,released low amounts of IL-12p70 and up-regulated IL-10 both at the mRNA and at the protein level. Upon activation with HGFMo,allogeneic CD4+CD25- T cells expressed the T regulatory (Treg)-associated transcription factor FoxP3,proliferated poorly,and released high levels of IL-10. Interestingly,blockade of surface immunoglobulin-like transcript 3 (ILT3) on HGFMo or neutralization of secreted IL-10 translated into partial restoration of T-cell proliferation. Secondary stimulation of HGFMo-primed CD4+ T cells with immunogenic DCs differentiated with granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 from monocytes of the same donor resulted in measurable T-cell proliferation. HGFMo-primed CD4+ T cells significantly inhibited the proliferation of naive CD4+CD25- T cells in a cell-contact-dependent manner. Finally,DNA microarray analysis revealed a unique gene-expression profile of HGF-activated monocytes. Collectively,our findings point to a novel role for HGF in the regulation of monocyte/DC functions that might be exploited therapeutically.
View Publication
产品号#:
09500
产品名:
BIT 9500血清替代物
Ma Y et al. (OCT 2006)
Blood 108 8 2726--35
SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice.
SALL4,a human homolog to Drosophila spalt,is a novel zinc finger transcriptional factor essential for development. We cloned SALL4 and its isoforms (SALL4A and SALL4B). Through immunohistochemistry and real-time reverse-transcription-polymerase chain reaction (RT-PCR),we demonstrated that SALL4 was constitutively expressed in human primary acute myeloid leukemia (AML,n = 81),and directly tested the leukemogenic potential of constitutive expression of SALL4 in a murine model. SALL4B transgenic mice developed myelodysplastic syndrome (MDS)-like features and subsequently AML that was transplantable. Increased apoptosis associated with dysmyelopoiesis was evident in transgenic mouse marrow and colony-formation (CFU) assays. Both isoforms could bind to beta-catenin and synergistically enhanced the Wnt/beta-catenin signaling pathway. Our data suggest that the constitutive expression of SALL4 causes MDS/AML,most likely through the Wnt/beta-catenin pathway. Our murine model provides a useful platform to study human MDS/AML transformation,as well as the Wnt/beta-catenin pathway's role in the pathogenesis of leukemia stem cells.
View Publication
产品号#:
03434
03444
产品名:
MethoCult™ GF M3434
MethoCult™ GF M3434
Kurita R et al. (SEP 2006)
Stem cells (Dayton,Ohio) 24 9 2014--22
Tal1/Scl gene transduction using a lentiviral vector stimulates highly efficient hematopoietic cell differentiation from common marmoset (Callithrix jacchus) embryonic stem cells.
The development of embryonic stem cell (ESC) therapies requires the establishment of efficient methods to differentiate ESCs into specific cell lineages. Here,we report the in vitro differentiation of common marmoset (CM) (Callithrix jacchus) ESCs into hematopoietic cells after exogenous gene transfer using vesicular stomatitis virus-glycoprotein-pseudotyped lentiviral vectors. We transduced hematopoietic genes,including tal1/scl,gata1,gata2,hoxB4,and lhx2,into CM ESCs. By immunochemical and morphological analyses,we demonstrated that overexpression of tal1/scl,but not the remaining genes,dramatically increased hematopoiesis of CM ESCs,resulting in multiple blood-cell lineages. Furthermore,flow cytometric analysis demonstrated that CD34,a hematopoietic stem/progenitor cell marker,was highly expressed in tal1/scl-overexpressing embryoid body cells. Similar results were obtained from three independent CM ESC lines. These results suggest that transduction of exogenous tal1/scl cDNA into ESCs is a promising method to induce the efficient differentiation of CM ESCs into hematopoietic stem/progenitor cells.
View Publication
产品号#:
03434
03444
04435
04445
产品名:
MethoCult™ GF M3434
MethoCult™ GF M3434
MethoCult™ H4435 Enriched
MethoCult™ H4435 Enriched
Wunderlich M et al. (SEP 2006)
Blood 108 5 1690--7
Human CD34+ cells expressing the inv(16) fusion protein exhibit a myelomonocytic phenotype with greatly enhanced proliferative ability.
The t(16:16) and inv(16) are associated with FAB M4Eo myeloid leukemias and result in fusion of the CBFB gene to the MYH11 gene (encoding smooth muscle myosin heavy chain [SMMHC]). Knockout of CBFbeta causes embryonic lethality due to lack of definitive hematopoiesis. Although knock-in of CBFB-MYH11 is not sufficient to cause disease,expression increases the incidence of leukemia when combined with cooperating events. Although mouse models are valuable tools in the study of leukemogenesis,little is known about the contribution of CBFbeta-SMMHC to human hematopoietic stem and progenitor cell self-renewal. We introduced the CBFbeta-MYH11 cDNA into human CD34+ cells via retroviral transduction. Transduced cells displayed an initial repression of progenitor activity but eventually dominated the culture,resulting in the proliferation of clonal populations for up to 7 months. Long-term cultures displayed a myelomonocytic morphology while retaining multilineage progenitor activity and engraftment in NOD/SCID-B2M-/- mice. Progenitor cells from long-term cultures showed altered expression of genes defining inv(16) identified in microarray studies of human patient samples. This system will be useful in examining the effects of CBFbeta-SMMHC on gene expression in the human preleukemic cell,in characterizing the effect of this oncogene on human stem cell biology,and in defining its contribution to the development of leukemia.
View Publication
产品号#:
04100
18056
18056RF
产品名:
MethoCult™ H4100
Griswold IJ et al. (AUG 2006)
Molecular and cellular biology 26 16 6082--93
Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib.
Kinase domain (KD) mutations of Bcr-Abl interfering with imatinib binding are the major mechanism of acquired imatinib resistance in patients with Philadelphia chromosome-positive leukemia. Mutations of the ATP binding loop (p-loop) have been associated with a poor prognosis. We compared the transformation potency of five common KD mutants in various biological assays. Relative to unmutated (native) Bcr-Abl,the ATP binding loop mutants Y253F and E255K exhibited increased transformation potency,M351T and H396P were less potent,and the performance of T315I was assay dependent. The transformation potency of Y253F and M351T correlated with intrinsic Bcr-Abl kinase activity,whereas the kinase activity of E255K,H396P,and T315I did not correlate with transforming capabilities,suggesting that additional factors influence transformation potency. Analysis of the phosphotyrosine proteome by mass spectroscopy showed differential phosphorylation among the mutants,a finding consistent with altered substrate specificity and pathway activation. Mutations in the KD of Bcr-Abl influence kinase activity and signaling in a complex fashion,leading to gain- or loss-of-function variants. The drug resistance and transformation potency of mutants may determine the outcome of patients on therapy with Abl kinase inhibitors.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒





 技术公告Identification of ALDH-Expressing Cancer Stem Cells Using ALDEFLUOR™
技术公告Identification of ALDH-Expressing Cancer Stem Cells Using ALDEFLUOR™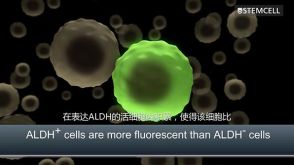

 沪公网安备31010102008431号
沪公网安备31010102008431号