Characterization of primitive hematopoietic cells from patients with dyskeratosis congenita.
Dyskeratosis congenita (DC) is an inherited bone marrow (BM) failure syndrome associated with mutations in telomerase genes and the acquisition of shortened telomeres in blood cells. To investigate the basis of the compromised hematopoiesis seen in DC,we analyzed cells from granulocyte colony-stimulating factor mobilized peripheral blood (mPB) collections from 5 members of a family with autosomal dominant DC with a hTERC mutation. Premobilization BM samples were hypocellular,and percentages of CD34(+) cells in marrow and mPB collections were significantly below values for age-matched controls in 4 DC subjects. Directly clonogenic cells,although present at normal frequencies within the CD34(+) subset,were therefore absolutely decreased. In contrast,even the frequency of long-term culture-initiating cells within the CD34(+) DC mPB cells was decreased,and the telomere lengths of these cells were also markedly reduced. Nevertheless,the different lineages of mature cells were produced in normal numbers in vitro. These results suggest that marrow failure in DC is caused by a reduction in the ability of hematopoietic stem cells to sustain their numbers due to telomere impairment rather than a qualitative defect in their commitment to specific lineages or in the ability of their lineage-restricted progeny to execute normal differentiation programs.
View Publication
产品号#:
04434
04444
09600
09650
18056
18056RF
产品名:
MethoCult™ H4434 Classic
MethoCult™ H4434 Classic
StemSpan™ SFEM
StemSpan™ SFEM
Iversen PO et al. (MAR 2010)
American journal of physiology. Regulatory,integrative and comparative physiology 298 3 R808--14
Separate mechanisms cause anemia in ischemic vs. nonischemic murine heart failure.
In ischemic congestive heart failure (CHF),anemia is associated with poor prognosis. Whether anemia develops in nonischemic CHF is uncertain. The hematopoietic inhibitors TNF-alpha and nitric oxide (NO) are activated in ischemic CHF. We examined whether mice with ischemic or nonischemic CHF develop anemia and whether TNF-alpha and NO are involved. We studied mice (n = 7-9 per group) with CHF either due to myocardial infarction (MI) or to overexpression of the Ca(2+)-binding protein calsequestrin (CSQ) or to induced cardiac disruption of the sarcoplasmic reticulum Ca(2+)-ATPase 2 gene (SERCA2 KO). Hematopoiesis was analyzed by colony formation of CD34(+) bone marrow cells. Hemoglobin concentration was 14.0 +/- 0.4 g/dl (mean +/- SD) in controls,while it was decreased to 10.1 +/- 0.4,9.7 +/- 0.4,and 9.6 +/- 0.3 g/dl in MI,CSQ,and SERCA2 KO,respectively (P textless 0.05). Colony numbers per 100,000 CD34(+) cells in the three CHF groups were reduced to 33 +/- 3 (MI),34 +/- 3 (CSQ),and 39 +/- 3 (SERCA2 KO) compared with 68 +/- 4 in controls (P textless 0.05). Plasma TNF-alpha nearly doubled in MI,and addition of anti-TNF-alpha antibody normalized colony formation. Inhibition of colony formation was completely abolished with blockade of endothelial NO synthase in CSQ and SERCA2 KO,but not in MI. In conclusion,the mechanism of anemia in CHF depends on the etiology of cardiac disease; whereas TNF-alpha impairs hematopoiesis in CHF following MI,NO inhibits blood cell formation in nonischemic murine CHF.
View Publication
产品号#:
产品名:
Ghiaur G et al. (APR 2008)
Blood 111 7 3313--21
Rac1 is essential for intraembryonic hematopoiesis and for the initial seeding of fetal liver with definitive hematopoietic progenitor cells.
Definitive hematopoietic stem and progenitor cells (HSCs/Ps) originating from the yolk sac and/or para-aorta-splanchno-pleura/aorta-gonad-mesonephros are hypothesized to colonize the fetal liver,but mechanisms involved are poorly defined. The Rac subfamily of Rho GTPases has been shown to play essential roles in HSC/P localization to the bone marrow following transplantation. Here,we study the role of Rac1 in HSC/P migration during ontogeny and seeding of fetal liver. Using a triple-transgenic approach,we have deleted Rac1 in HSCs/Ps during very early embryonic development. Without Rac1,there was a decrease in circulating HSCs/Ps in the blood of embryonic day (E) 10.5 embryos,while yolk sac definitive hematopoiesis was quantitatively normal. Intraembryonic hematopoiesis was significantly impaired in Rac1-deficient embryos,culminating with absence of intra-aortic clusters and fetal liver hematopoiesis. At E10.5,Rac1-deficient HSCs/Ps displayed decreased transwell migration and impaired inter-action with the microenvironment in migration-dependent assays. These data suggest that Rac1 plays an important role in HSC/P migration during embryonic development and is essential for the emergence of intraembryonic hematopoiesis.
View Publication
产品号#:
03134
09600
09650
产品名:
MethoCult™ M3134
StemSpan™ SFEM
StemSpan™ SFEM
Qiu C et al. (FEB 2008)
Blood 111 4 2400--8
Globin switches in yolk sac-like primitive and fetal-like definitive red blood cells produced from human embryonic stem cells.
We have previously shown that coculture of human embryonic stem cells (hESCs) for 14 days with immortalized fetal hepatocytes yields CD34(+) cells that can be expanded in serum-free liquid culture into large numbers of megaloblastic nucleated erythroblasts resembling yolk sac-derived cells. We show here that these primitive erythroblasts undergo a switch in hemoglobin (Hb) composition during late terminal erythroid maturation with the basophilic erythroblasts expressing predominantly Hb Gower I (zeta(2)epsilon(2)) and the orthochromatic erythroblasts hemoglobin Gower II (alpha(2)epsilon(2)). This suggests that the switch from Hb Gower I to Hb Gower II,the first hemoglobin switch in humans is a maturation switch not a lineage switch. We also show that extending the coculture of the hESCs with immortalized fetal hepatocytes to 35 days yields CD34(+) cells that differentiate into more developmentally mature,fetal liver-like erythroblasts,that are smaller,express mostly fetal hemoglobin,and can enucleate. We conclude that hESC-derived erythropoiesis closely mimics early human development because the first 2 human hemoglobin switches are recapitulated,and because yolk sac-like and fetal liver-like cells are sequentially produced. Development of a method that yields erythroid cells with an adult phenotype remains necessary,because the most mature cells that can be produced with current systems express less than 2% adult beta-globin mRNA.
View Publication
产品号#:
09600
09650
18056
18056RF
产品名:
StemSpan™ SFEM
StemSpan™ SFEM
Sauer AV et al. (OCT 2009)
Blood 114 15 3216--26
ADA-deficient SCID is associated with a specific microenvironment and bone phenotype characterized by RANKL/OPG imbalance and osteoblast insufficiency.
Adenosine deaminase (ADA) deficiency is a disorder of the purine metabolism leading to combined immunodeficiency and systemic alterations,including skeletal abnormalities. We report that ADA deficiency in mice causes a specific bone phenotype characterized by alterations of structural properties and impaired mechanical competence. These alterations are the combined result of an imbalanced receptor activator of nuclear factor-kappaB ligand (RANKL)/osteoprotegerin axis,causing decreased osteoclastogenesis and an intrinsic defect of osteoblast function with subsequent low bone formation. In vitro,osteoblasts lacking ADA displayed an altered transcriptional profile and growth reduction. Furthermore,the bone marrow microenvironment of ADA-deficient mice showed a reduced capacity to support in vitro and in vivo hematopoiesis. Treatment of ADA-deficient neonatal mice with enzyme replacement therapy,bone marrow transplantation,or gene therapy resulted in full recovery of the altered bone parameters. Remarkably,untreated ADA-severe combined immunodeficiency patients showed a similar imbalance in RANKL/osteoprotegerin levels alongside severe growth retardation. Gene therapy with ADA-transduced hematopoietic stem cells increased serum RANKL levels and children's growth. Our results indicate that the ADA metabolism represents a crucial modulatory factor of bone cell activities and remodeling.
View Publication
产品号#:
13056
产品名:
Twu Y-C et al. (MAR 2010)
Blood 115 12 2491--9
Phosphorylation status of transcription factor C/EBPalpha determines cell-surface poly-LacNAc branching (I antigen) formation in erythropoiesis and granulopoiesis.
The cell-surface straight and branched repeats of N-acetyllactosamine (LacNAc) units,called poly-LacNAc chains,characterize the histo-blood group i and I antigens,respectively. The transition of straight to branched poly-LacNAc chain (i to I) is determined by the I locus,which expresses 3 IGnT transcripts,IGnTA,IGnTB,and IGnTC. Our previous investigation demonstrated that the i-to-I transition in erythroid differentiation is regulated by the transcription factor CCAAT/enhancer binding protein alpha (C/EBPalpha). In the present investigation,the K-562 cell line was used as a model to show that the i-to-I transition is determined by the phosphorylation status of the C/EBPalpha Ser-21 residue,with dephosphorylated C/EBPalpha Ser-21 stimulating the transcription of the IGnTC gene,consequently resulting in I branching. Results from studies using adult erythropoietic and granulopoietic progenitor cells agreed with those derived using the K-562 cell model,with lentiviral expression of C/EBPalpha in CD34(+) hematopoietic cells demonstrating that the dephosphorylated form of C/EBPalpha Ser-21 induced the expression of I antigen,granulocytic CD15,and also erythroid CD71 antigens. Taken together,these results demonstrate that the regulation of poly-LacNAc branching (I antigen) formation in erythropoiesis and granulopoiesis share a common mechanism,with dephosphorylation of the Ser-21 residue on C/EBPalpha playing the critical role.
View Publication
产品号#:
02532
02832
02615
02855
09600
09650
产品名:
StemSpan™ SFEM
StemSpan™ SFEM
Thein SL et al. (JUL 2007)
Proceedings of the National Academy of Sciences of the United States of America 104 27 11346--51
Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults.
Individual variation in fetal hemoglobin (HbF,alpha(2)gamma(2)) response underlies the remarkable diversity in phenotypic severity of sickle cell disease and beta thalassemia. HbF levels and HbF-associated quantitative traits (e.g.,F cell levels) are highly heritable. We have previously mapped a major quantitative trait locus (QTL) controlling F cell levels in an extended Asian-Indian kindred with beta thalassemia to a 1.5-Mb interval on chromosome 6q23,but the causative gene(s) are not known. The QTL encompasses several genes including HBS1L,a member of the GTP-binding protein family that is expressed in erythroid progenitor cells. In this high-resolution association study,we have identified multiple genetic variants within and 5' to HBS1L at 6q23 that are strongly associated with F cell levels in families of Northern European ancestry (P = 10(-75)). The region accounts for 17.6% of the F cell variance in northern Europeans. Although mRNA levels of HBS1L and MYB in erythroid precursors grown in vitro are positively correlated,only HBS1L expression correlates with high F cell alleles. The results support a key role for the HBS1L-related genetic variants in HbF control and illustrate the biological complexity of the mechanism of 6q QTL as a modifier of fetal hemoglobin levels in the beta hemoglobinopathies.
View Publication
产品号#:
09600
09650
产品名:
StemSpan™ SFEM
StemSpan™ SFEM
Baba Y et al. (AUG 2006)
Journal of immunology (Baltimore,Md. : 1950) 177 4 2294--303
Constitutively active beta-catenin promotes expansion of multipotent hematopoietic progenitors in culture.
This study was designed to investigate one component of the Wnt/beta-catenin signaling pathway that has been implicated in stem cell self-renewal. Retroviral-mediated introduction of stable beta-catenin to primitive murine bone marrow cells allowed the expansion of multipotential c-Kit(low)Sca-1(low/-)CD19(-) CD11b/Mac-1(-)Flk-2(-)CD43(+)AA4.1(+)NK1.1(-)CD3(-)CD11c(-)Gr-1(-)CD45R/B220(+) cells in the presence of stromal cells and cytokines. They generated myeloid,T,and B lineage lymphoid cells in culture,but had no T lymphopoietic potential when transplanted. Stem cell factor and IL-6 were found to be minimal requirements for long-term,stromal-free propagation,and a beta-catenin-transduced cell line was maintained for 5 mo with these defined conditions. Although multipotential and responsive to many normal stimuli in culture,it was unable to engraft several types of irradiated recipients. These findings support previous studies that have implicated the canonical Wnt pathway signaling in regulation of multipotent progenitors. In addition,we demonstrate how it may be experimentally manipulated to generate valuable cell lines.
View Publication
产品号#:
03434
03444
产品名:
MethoCult™ GF M3434
MethoCult™ GF M3434
Nakagawa M et al. (NOV 2006)
Blood 108 10 3329--34
AML1/Runx1 rescues Notch1-null mutation-induced deficiency of para-aortic splanchnopleural hematopoiesis.
The Notch1-RBP-Jkappa and the transcription factor Runx1 pathways have been independently shown to be indispensable for the establishment of definitive hematopoiesis. Importantly,expression of Runx1 is down-regulated in the para-aortic splanchnopleural (P-Sp) region of Notch1- and Rbpsuh-null mice. Here we demonstrate that Notch1 up-regulates Runx1 expression and that the defective hematopoietic potential of Notch1-null P-Sp cells is successfully rescued in the OP9 culture system by retroviral transfer of Runx1. We also show that Hes1,a known effector of Notch signaling,potentiates Runx1-mediated transactivation. Together with the recent findings in zebrafish,Runx1 is postulated to be a cardinal down-stream mediator of Notch signaling in hematopoietic development throughout vertebrates. Our findings also suggest that Notch signaling may modulate both expression and transcriptional activity of Runx1.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒





 技术公告Identification of ALDH-Expressing Cancer Stem Cells Using ALDEFLUOR™
技术公告Identification of ALDH-Expressing Cancer Stem Cells Using ALDEFLUOR™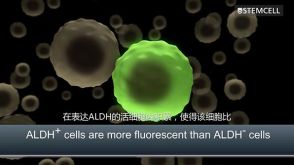

 沪公网安备31010102008431号
沪公网安备31010102008431号