Pirson L et al. (JUL 2006)
Stem cells (Dayton,Ohio) 24 7 1814--21
Despite inhibition of hematopoietic progenitor cell growth in vitro, the tyrosine kinase inhibitor imatinib does not impair engraftment of human CD133+ cells into NOD/SCIDbeta2mNull mice.
There is potential interest for combining allogeneic hematopoietic cell transplantation (HCT),and particularly allogeneic HCT with a nonmyeloablative regimen,to the tyrosine kinase inhibitor imatinib (Glivec; Novartis,Basel,Switzerland,http://www.novartis.com) in order to maximize anti-leukemic activity against Philadelphia chromosome-positive leukemias. However,because imatinib inhibits c-kit,the stem cell factor receptor,it could interfere with bone marrow engraftment. In this study,we examined the impact of imatinib on normal progenitor cell function. Imatinib decreased the colony-forming capacity of mobilized peripheral blood human CD133(+) cells but not that of long-term culture-initiating cells. Imatinib also decreased the proliferation of cytokine-stimulated CD133(+) cells but did not induce apoptosis of these cells. Expression of very late antigen (VLA)-4,VLA-5,and CXCR4 of CD133(+) cells was not modified by imatinib,but imatinib decreased the ability of CD133(+) cells to migrate. Finally,imatinib did not decrease engraftment of CD133(+) cells into irradiated nonobese diabetic/severe combined immunodeficient/beta2m(null) mice conditioned with 3 or 1 Gy total body irradiation. In summary,our results suggest that,despite inhibition of hematopoietic progenitor cell growth in vitro,imatinib does not interfere with hematopoietic stem cell engraftment.
View Publication
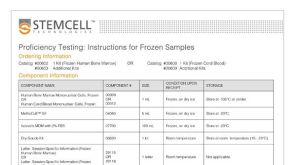
产品号#:
05150
04435
04445
04960
04902
04900
04961
04901
04963
04962
04970
04971
产品名:
MyeloCult™ H5100
MethoCult™ H4435 Enriched
MethoCult™ H4435 Enriched
MegaCult™-C胶原和无细胞因子培养基
胶原蛋白溶液
MegaCult™-C无细胞因子培养基
MegaCult™-C胶原和含细胞因子培养基
MegaCult™-C含细胞因子培养基
双室载玻片套件
MegaCult™-C CFU-Mk染色试剂盒
MegaCult™-C无细胞因子全套试剂盒
MegaCult™-C含细胞因子全套试剂盒
Goel A et al. (MAY 2006)
Blood 107 10 4063--70
Synergistic activity of the proteasome inhibitor PS-341 with non-myeloablative 153-Sm-EDTMP skeletally targeted radiotherapy in an orthotopic model of multiple myeloma.
Multiple myeloma is a highly radiosensitive skeletal malignancy,but bone-seeking radionuclides have not yet found their place in disease management. We previously reported that the proteasome inhibitor PS-341 selectively sensitizes myeloma cells to the lethal effects of ionizing radiation. To extend these observations to an in vivo model,we combined PS-341 with the bone-seeking radionuclide 153-Sm-EDTMP. In vitro clonogenic assays demonstrated synergistic killing of myeloma cells exposed to both PS-341 and 153-Sm-EDTMP. Using the orthotopic,syngeneic 5TGM1 myeloma model,the median survivals of mice treated with saline,2 doses of PS-341 (0.5 mg/kg),or a single nonmyeloablative dose of 153-Sm-EDTMP (22.5 MBq) were 21,22,and 28 days,respectively. In contrast,mice treated with combination therapy comprising 2 doses of PS-341 (0.5 mg/kg),1 day prior to and 1 day following 153-Sm-EDTMP (22.5 MBq) showed a significantly prolonged median survival of 49 days (P textless .001). In addition to prolonged survival,this treatment combination yielded reduced clonogenicity of bone marrow-resident 5TGM1 cells,reduced serum myeloma-associated paraprotein levels,and better preservation of bone mineral density. Myelosuppression,determined by peripheral blood cell counts and clonogenicity assays of hematopoietic progenitors,did not differ between animals treated with 153-Sm-EDTMP alone versus those treated with the combination of PS-341 plus 153-Sm-EDTMP. PS-341 is a potent,selective in vivo radiosensitizer that may substantially affect the efficacy of skeletal-targeted radiotherapy in multiple myeloma.
View Publication
产品号#:
04236
产品名:
MethoCult™ SF H4236
Senatus PB et al. (JAN 2006)
Molecular cancer therapeutics 5 1 20--8
Restoration of p53 function for selective Fas-mediated apoptosis in human and rat glioma cells in vitro and in vivo by a p53 COOH-terminal peptide.
We have shown that a COOH-terminal peptide of p53 (amino acids 361-382,p53p),linked to the truncated homeobox domain of Antennapedia (Ant) as a carrier for transduction,induced rapid apoptosis in human premalignant and malignant cell lines. Here,we report that human and rat glioma lines containing endogenous mutant p53 or wild-type (WT) p53 were induced into apoptosis by exposure to this peptide called p53p-Ant. The peptide was comparatively nontoxic to proliferating nonmalignant human and rat glial cell lines containing WT p53 and proliferating normal human peripheral marrow blood stem cells. Degree of sensitivity to the peptide correlated directly with the level of endogenous p53 expression and mutant p53 conformation. Apoptosis induction by p53p-Ant was quantitated by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay and Annexin V staining in human glioma cells in vitro and in a syngeneic orthotopic 9L glioma rat model using convection-enhanced delivery in vivo. The mechanism of cell death by this peptide was solely through the Fas extrinsic apoptotic pathway. p53p-Ant induced a 3-fold increase in extracellular membrane Fas expression in glioma cells but no significant increase in nonmalignant glial cells. These data suggest that p53 function for inducing Fas-mediated apoptosis in gliomas,which express sufficient quantities of endogenous mutant or WT p53,may be restored or activated,respectively,by a cell-permeable peptide derived from the p53 COOH-terminal regulatory domain (p53p-Ant). p53p-Ant may serve as a prototypic model for the development of new anticancer agents with unique selectivity for glioma cancer cells and it can be successfully delivered in vivo into a brain tumor by a convection-enhanced delivery system,which circumvents the blood-brain barrier.
View Publication
Cohen-Haguenauer O et al. (FEB 2006)
Proceedings of the National Academy of Sciences of the United States of America 103 7 2340--5
In vivo repopulation ability of genetically corrected bone marrow cells from Fanconi anemia patients.
Fanconi anemia (FA) is a rare inherited genomic instability syndrome representing one of the best examples of hematopoietic stem cell deficiency. Although FA might be an excellent candidate for bone marrow (BM) genetic correction ex vivo,knockout animal models are not sufficient to guide preclinical steps,and gene therapy attempts have proven disappointing so far. Contributing to these poor results is a characteristic and dramatic early BM-cells die-off when placed in culture. We show here that human primary FA BM cell survival can be ameliorated by using specific culture conditions that limit oxidative stress. When coupled with retrovirus-mediated transfer of the main complementation group FANCA-cDNA,we could achieve long-term reconstitution of the stem cell compartment both in vitro and in vivo. Gene-corrected BM cultures grew for textgreater120 days,and after cultured cell transplantation into NOD/SCID mice,clonogenic human cells carrying the FANCA transgene could be detected 6 months after transduction. By comparison,untransduced cells died in culture by 15 days. Of necessity for ethical reasons,experiments were conducted on a very limited number of primary BM cells. By using low cytokine regimen and conditions matching regulatory requirements,a contingent of gene-corrected cells slowly emerges with an unmet potential for in vivo engraftment. Future therapeutic applications of stem cells might be expanding from these data. In addition,we provide a model of gene-corrected human primary cell growth that carries the potential to better delineate the combined role of both DNA damage and oxidative stress in the pathogenesis of FA.
View Publication
产品号#:
04436
产品名:
MethoCult™ SF H4436
Rutella S et al. (JUL 2006)
Blood 108 1 218--27
Hepatocyte growth factor favors monocyte differentiation into regulatory interleukin (IL)-10++IL-12low/neg accessory cells with dendritic-cell features.
Several hematopoietic growth factors,including interleukin-10 (IL-10) and transforming growth factor-beta1 (TGF-beta1),promote the differentiation of tolerogenic dendritic cells (DCs). Hepatocyte growth factor (HGF) is a pleiotropic cytokine whose effects on human DC differentiation and function have not been investigated. Monocytes cultured with HGF (HGFMo) differentiated into accessory cells with DC-like morphology,released low amounts of IL-12p70 and up-regulated IL-10 both at the mRNA and at the protein level. Upon activation with HGFMo,allogeneic CD4+CD25- T cells expressed the T regulatory (Treg)-associated transcription factor FoxP3,proliferated poorly,and released high levels of IL-10. Interestingly,blockade of surface immunoglobulin-like transcript 3 (ILT3) on HGFMo or neutralization of secreted IL-10 translated into partial restoration of T-cell proliferation. Secondary stimulation of HGFMo-primed CD4+ T cells with immunogenic DCs differentiated with granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 from monocytes of the same donor resulted in measurable T-cell proliferation. HGFMo-primed CD4+ T cells significantly inhibited the proliferation of naive CD4+CD25- T cells in a cell-contact-dependent manner. Finally,DNA microarray analysis revealed a unique gene-expression profile of HGF-activated monocytes. Collectively,our findings point to a novel role for HGF in the regulation of monocyte/DC functions that might be exploited therapeutically.
View Publication
SALL4, a novel oncogene, is constitutively expressed in human acute myeloid leukemia (AML) and induces AML in transgenic mice.
SALL4,a human homolog to Drosophila spalt,is a novel zinc finger transcriptional factor essential for development. We cloned SALL4 and its isoforms (SALL4A and SALL4B). Through immunohistochemistry and real-time reverse-transcription-polymerase chain reaction (RT-PCR),we demonstrated that SALL4 was constitutively expressed in human primary acute myeloid leukemia (AML,n = 81),and directly tested the leukemogenic potential of constitutive expression of SALL4 in a murine model. SALL4B transgenic mice developed myelodysplastic syndrome (MDS)-like features and subsequently AML that was transplantable. Increased apoptosis associated with dysmyelopoiesis was evident in transgenic mouse marrow and colony-formation (CFU) assays. Both isoforms could bind to beta-catenin and synergistically enhanced the Wnt/beta-catenin signaling pathway. Our data suggest that the constitutive expression of SALL4 causes MDS/AML,most likely through the Wnt/beta-catenin pathway. Our murine model provides a useful platform to study human MDS/AML transformation,as well as the Wnt/beta-catenin pathway's role in the pathogenesis of leukemia stem cells.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒









 沪公网安备31010102008431号
沪公网安备31010102008431号