Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis.
In order to investigate the biologic processes underlying and resulting from the megakaryocytic hyperplasia that characterizes idiopathic myelofibrosis (IMF),peripheral blood CD34+ cells isolated from patients with IMF,polycythemia vera (PV),and G-CSF-mobilized healthy volunteers were cultured in the presence of stem cell factor and thrombopoietin. IMF CD34+ cells generated 24-fold greater numbers of megakaryocytes (MKs) than normal CD34+ cells. IMF MKs were also shown to have a delayed pattern of apoptosis and to overexpress the antiapoptotic protein bcl-xL. MK hyperplasia in IMF is,therefore,likely a consequence of both the increased ability of IMF progenitor cells to generate MKs and a decreased rate of MK apoptosis. Media conditioned (CM) by CD61+ cells generated in vitro from CD34+ cells were then assayed for the levels of growth factors and proteases. Higher levels of transforming growth factor-beta (TGF-beta) and active matrix metalloproteinase-9 (MMP9) were observed in media conditioned with IMF CD61+ cells than normal or PV CD61+ cells. Both normal and IMF CD61+ cells produced similar levels of VEGF. MK-derived TGF-B and MMP-9,therefore,likely contribute to the development of many pathological epiphenomena associated with IMF.
View Publication
Seo J-H et al. (SEP 2010)
Cancer research 70 18 7325--35
A specific need for CRKL in p210BCR-ABL-induced transformation of mouse hematopoietic progenitors.
CRKL (CRK-like) is an adapter protein predominantly phosphorylated in cells that express the tyrosine kinase p210(BCR-ABL),the fusion product of a (9;22) chromosomal translocation causative for chronic myeloid leukemia. It has been unclear,however,whether CRKL plays a functional role in p210(BCR-ABL) transformation. Here,we show that CRKL is required for p210(BCR-ABL) to support interleukin-3-independent growth of myeloid progenitor cells and long-term outgrowth of B-lymphoid cells from fetal liver-derived hematopoietic progenitor cells. Furthermore,a synthetic phosphotyrosyl peptide that binds to the CRKL SH2 domain with high affinity blocks association of endogenous CRKL with the p210(BCR-ABL) complex and reduces c-MYC levels in K562 human leukemic cells as well as in mouse hematopoietic cells transformed by p210(BCR-ABL) or the imatinib-resistant mutant T315I. These results indicate that the function of CRKL as an adapter protein is essential for p210(BCR-ABL)-induced transformation.
View Publication
Agosti V et al. (MAR 2004)
The Journal of experimental medicine 199 6 867--78
Critical role for Kit-mediated Src kinase but not PI 3-kinase signaling in pro T and pro B cell development.
The Kit receptor functions in hematopoiesis,lymphocyte development,gastrointestinal tract motility,melanogenesis,and gametogenesis. To investigate the roles of different Kit signaling pathways in vivo,we have generated knock-in mice in which docking sites for PI 3-kinase (KitY719) or Src kinase (KitY567) have been mutated. Whereas steady-state hematopoiesis is normal in KitY719F/Y719F and KitY567F/Y567F mice,lymphopoiesis is affected differentially. The KitY567F mutation,but not the KitY719F mutation,blocks pro T cell and pro B cell development in an age-dependent manner. Thus,the Src family kinase,but not the PI 3-kinase docking site in Kit,mediates a critical signal for lymphocyte development. In agreement with these results,treatment of normal mice with the Kit tyrosine kinase inhibitor imatinib (Gleevec) leads to deficits in pro T and pro B cell development,similar to those seen in KitY567F/Y567F and KitW/W mice. The two mutations do not affect embryonic gametogenesis but the KitY719F mutation blocks spermatogenesis at the spermatogonial stages and in contrast the KitY567F mutation does not affect this process. Therefore,Kit-mediated PI 3-kinase signaling and Src kinase family signaling is highly specific for different cellular contexts in vivo.
View Publication
Activation-induced cytidine deaminase deficiency accelerates autoimmune diabetes in NOD mice.
B cells play an important role in type 1 diabetes (T1D) development. However,the role of B cell activation-induced cytidine deaminase (AID) in diabetes development is not clear. We hypothesized that AID is important in the immunopathogenesis of T1D. To test this hypothesis,we generated AID-deficient (AID-/-) NOD mice. We found that AID-/-NOD mice developed accelerated T1D,with worse insulitis and high levels of anti-insulin autoantibody in the circulation. Interestingly,neither maternal IgG transferred through placenta,nor IgA transferred through milk affected the accelerated diabetes development. AID-/-NOD mice showed increased activation and proliferation of B and T cells. We found enhanced T-B cell interactions in AID-/-NOD mice,with increased T-bet and IFN-γ expression in CD4+ T cells in the presence of AID-/- B cells. Moreover,excessive lymphoid expansion was observed in AID-/-NOD mice. Importantly,antigen-specific BDC2.5 CD4+ T cells caused more rapid onset of diabetes when cotransferred with AID-/- B cells than when cotransferred with AID+/+ B cells. Thus,our study provides insights into the role of AID in T1D. Our data also suggest that AID is a negative regulator of immune tolerance and ablation of AID can lead to exacerbated islet autoimmunity and accelerated T1D development.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




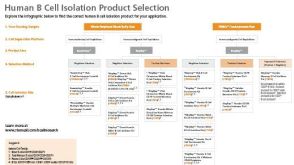
 实验方案Optimizing Delivery Efficiency with Fluorescent Dextran Using the CellPore™ Transfection System
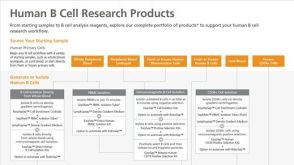
实验方案Optimizing Delivery Efficiency with Fluorescent Dextran Using the CellPore™ Transfection System



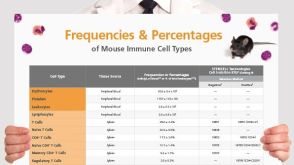 挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice
挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice
 沪公网安备31010102008431号
沪公网安备31010102008431号