Loss of p19Arf in a Rag1(-/-) B-cell precursor population initiates acute B-lymphoblastic leukemia.
In human B-acute lymphoblastic leukemia (B-ALL),RAG1-induced genomic alterations are important for disease progression. However,given that biallelic loss of the RAG1 locus is observed in a subset of cases,RAG1's role in the development of B-ALL remains unclear. We chose a p19Arf(-/-)Rag1(-/-) mouse model to confirm the previously published results concerning the contribution of CDKN2A (p19ARF /INK4a) and RAG1 copy number alterations in precursor B cells to the initiation and/or progression to B-acute lymphoblastic leukemia (B-ALL). In this murine model,we identified a new,Rag1-independent leukemia-initiating mechanism originating from a Sca1(+)CD19(+) precursor cell population and showed that Notch1 expression accelerates the cells' self-renewal capacity in vitro. In human RAG1-deficient BM,a similar CD34(+)CD19(+) population expressed p19ARF. These findings suggest that combined loss of p19Arf and Rag1 results in B-cell precursor leukemia in mice and may contribute to the progression of precursor B-ALL in humans.
View Publication
产品号#:
产品名:
Zan H et al. (JAN 2011)
Molecular immunology 48 4 610--22
Endonuclease G plays a role in immunoglobulin class switch DNA recombination by introducing double-strand breaks in switch regions.
Immunoglobulin (Ig) class switch DNA recombination (CSR) is the crucial mechanism diversifying the biological effector functions of antibodies. Generation of double-strand DNA breaks (DSBs),particularly staggered DSBs,in switch (S) regions of the upstream and downstream CH genes involved in the specific recombination process is an absolute requirement for CSR. Staggered DSBs would be generated through deamination of dCs on opposite DNA strands by activation-induced cytidine deaminase (AID),subsequent dU deglycosylation by uracil DNA glycosylase (Ung) and abasic site nicking by apurinic/apyrimidic endonuclease. However,consistent with the findings that significant amounts of DSBs can be detected in the IgH locus in the absence of AID or Ung,we have shown in human and mouse B cells that AID generates staggered DSBs not only by cleaving intact double-strand DNA,but also by processing blunt DSB ends generated in an AID-independent fashion. How these AID-independent DSBs are generated is still unclear. It is possible that S region DNA may undergo AID-independent cleavage by structure-specific nucleases,such as endonuclease G (EndoG). EndoG is an abundant nuclease in eukaryotic cells. It cleaves single and double-strand DNA,primarily at dG/dC residues,the preferential sites of DSBs in S region DNA. We show here that EndoG can localize to the nucleus of B cells undergoing CSR and binds to S region DNA,as shown by specific chromatin immunoprecipitation assays. Using knockout EndoG(-/-) mice and EndoG(-/-) B cells,we found that EndoG deficiency resulted in a two-fold reduction in CSR in vivo and in vitro,as demonstrated by reduced cell surface IgG1,IgG2a,IgG3 and IgA,reduced secreted IgG1,reduced circle Iγ1-Cμ,Iγ3-Cμ,Iɛ-Cμ,Iα-Cμ transcripts,post-recombination Iμ-Cγ1,Iμ-Cγ3,Iμ-Cɛ and Iμ-Cα transcripts. In addition to reduced CSR,EndoG(-/-) mice showed a significantly altered spectrum of mutations in IgH J(H)-iEμ DNA. Impaired CSR in EndoG(-/-) B cells did not stem from altered B cell proliferation or apoptosis. Rather,it was associated with significantly reduced frequency of DSBs. Thus,our findings determine a role for EndoG in the generation of S region DSBs and CSR.
View Publication
产品号#:
19754
19754RF
产品名:
Ciurea SO et al. (AUG 2007)
Blood 110 3 986--93
Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis.
In order to investigate the biologic processes underlying and resulting from the megakaryocytic hyperplasia that characterizes idiopathic myelofibrosis (IMF),peripheral blood CD34+ cells isolated from patients with IMF,polycythemia vera (PV),and G-CSF-mobilized healthy volunteers were cultured in the presence of stem cell factor and thrombopoietin. IMF CD34+ cells generated 24-fold greater numbers of megakaryocytes (MKs) than normal CD34+ cells. IMF MKs were also shown to have a delayed pattern of apoptosis and to overexpress the antiapoptotic protein bcl-xL. MK hyperplasia in IMF is,therefore,likely a consequence of both the increased ability of IMF progenitor cells to generate MKs and a decreased rate of MK apoptosis. Media conditioned (CM) by CD61+ cells generated in vitro from CD34+ cells were then assayed for the levels of growth factors and proteases. Higher levels of transforming growth factor-beta (TGF-beta) and active matrix metalloproteinase-9 (MMP9) were observed in media conditioned with IMF CD61+ cells than normal or PV CD61+ cells. Both normal and IMF CD61+ cells produced similar levels of VEGF. MK-derived TGF-B and MMP-9,therefore,likely contribute to the development of many pathological epiphenomena associated with IMF.
View Publication
产品号#:
09600
09650
产品名:
StemSpan™ SFEM
StemSpan™ SFEM
Walker WE et al. (OCT 2006)
Journal of immunology (Baltimore,Md. : 1950) 177 8 5307--16
Absence of innate MyD88 signaling promotes inducible allograft acceptance.
Prior experimental strategies to induce transplantation tolerance have focused largely on modifying adaptive immunity. However,less is known concerning the role of innate immune signaling in the induction of transplantation tolerance. Using a highly immunogenic murine skin transplant model that resists transplantation tolerance induction when innate immunity is preserved,we show that absence of MyD88,a key innate Toll like receptor signal adaptor,abrogates this resistance and facilitates inducible allograft acceptance. In our model,absence of MyD88 impairs inflammatory dendritic cell responses that reduce T cell activation. This effect increases T cell susceptibility to suppression mediated by CD4+ CD25+ regulatory T cells. Therefore,this study provides evidence that absence of MyD88 promotes inducible allograft acceptance and implies that inhibiting innate immunity may be a potential,clinically relevant strategy to facilitate transplantation tolerance.
View Publication
产品号#:
18758
18758RF
18768
18768RF
19752
19752RF
19753
19753RF
产品名:
Irish JM et al. (AUG 2006)
Journal of immunology (Baltimore,Md. : 1950) 177 3 1581--9
Kinetics of B cell receptor signaling in human B cell subsets mapped by phosphospecific flow cytometry.
Differences in BCR signaling may govern outcomes as diverse as proliferation and cell death. We profiled BCR signaling kinetics in subsets of primary human B cells using flow cytometry. In the predominant population expressing IgM,BCR cross-linking led to a quick burst of Syk,ERK1/2,and p38 signaling. In contrast,IgG B cells sustained higher per-cell ERK1/2 phosphorylation over time. This dichotomy suggested a mechanism for dampening signals transmitted by IgM. Regulatory phosphatase activity in IgM B cells was BCR-mediated and initiated more slowly than kinase activity. This BCR-mediated phosphatase activity was sensitive to inhibition by H(2)O(2) and required to attenuate IgM BCR signaling. These results provide the first kinetic maps of BCR signaling in primary human B cell subsets and enable new studies of signaling in B cell disorders,such as autoimmunity and cancer.
View Publication
Seo J-H et al. (SEP 2010)
Cancer research 70 18 7325--35
A specific need for CRKL in p210BCR-ABL-induced transformation of mouse hematopoietic progenitors.
CRKL (CRK-like) is an adapter protein predominantly phosphorylated in cells that express the tyrosine kinase p210(BCR-ABL),the fusion product of a (9;22) chromosomal translocation causative for chronic myeloid leukemia. It has been unclear,however,whether CRKL plays a functional role in p210(BCR-ABL) transformation. Here,we show that CRKL is required for p210(BCR-ABL) to support interleukin-3-independent growth of myeloid progenitor cells and long-term outgrowth of B-lymphoid cells from fetal liver-derived hematopoietic progenitor cells. Furthermore,a synthetic phosphotyrosyl peptide that binds to the CRKL SH2 domain with high affinity blocks association of endogenous CRKL with the p210(BCR-ABL) complex and reduces c-MYC levels in K562 human leukemic cells as well as in mouse hematopoietic cells transformed by p210(BCR-ABL) or the imatinib-resistant mutant T315I. These results indicate that the function of CRKL as an adapter protein is essential for p210(BCR-ABL)-induced transformation.
View Publication
Agosti V et al. (MAR 2004)
The Journal of experimental medicine 199 6 867--78
Critical role for Kit-mediated Src kinase but not PI 3-kinase signaling in pro T and pro B cell development.
The Kit receptor functions in hematopoiesis,lymphocyte development,gastrointestinal tract motility,melanogenesis,and gametogenesis. To investigate the roles of different Kit signaling pathways in vivo,we have generated knock-in mice in which docking sites for PI 3-kinase (KitY719) or Src kinase (KitY567) have been mutated. Whereas steady-state hematopoiesis is normal in KitY719F/Y719F and KitY567F/Y567F mice,lymphopoiesis is affected differentially. The KitY567F mutation,but not the KitY719F mutation,blocks pro T cell and pro B cell development in an age-dependent manner. Thus,the Src family kinase,but not the PI 3-kinase docking site in Kit,mediates a critical signal for lymphocyte development. In agreement with these results,treatment of normal mice with the Kit tyrosine kinase inhibitor imatinib (Gleevec) leads to deficits in pro T and pro B cell development,similar to those seen in KitY567F/Y567F and KitW/W mice. The two mutations do not affect embryonic gametogenesis but the KitY719F mutation blocks spermatogenesis at the spermatogonial stages and in contrast the KitY567F mutation does not affect this process. Therefore,Kit-mediated PI 3-kinase signaling and Src kinase family signaling is highly specific for different cellular contexts in vivo.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒







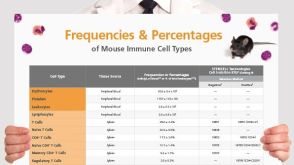 挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice
挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice
 沪公网安备31010102008431号
沪公网安备31010102008431号