Biologic and genetic characterization of the novel amyloidogenic lambda light chain-secreting human cell lines, ALMC-1 and ALMC-2.
Primary systemic amyloidosis (AL) is a rare monoclonal plasma cell (PC) disorder characterized by the deposition of misfolded immunoglobulin (Ig) light chains (LC) in vital organs throughout the body. To our knowledge,no cell lines have ever been established from AL patients. Here we describe the establishment of the ALMC-1 and ALMC-2 cell lines from an AL patient. Both cell lines exhibit a PC phenotype and display cytokine-dependent growth. Using a comprehensive genetic approach,we established the genetic relationship between the cell lines and the primary patient cells,and we were also able to identify new genetic changes accompanying tumor progression that may explain the natural history of this patient's disease. Importantly,we demonstrate that free lambda LC secreted by both cell lines contained a beta structure and formed amyloid fibrils. Despite absolute Ig LC variable gene sequence identity,the proteins show differences in amyloid formation kinetics that are abolished by the presence of Na(2)SO(4). The formation of amyloid fibrils from these naturally secreting human LC cell lines is unprecedented. Moreover,these cell lines will provide an invaluable tool to better understand AL,from the combined perspectives of amyloidogenic protein structure and amyloid formation,genetics,and cell biology.
View Publication
产品号#:
18357
18357RF
21000
20119
20155
18387
18387RF
产品名:
RoboSep™- S
RoboSep™ 吸头组件抛光剂
RoboSep™分选管套装(9个塑料管)
Douglas KB et al. (JUL 2009)
Genes and immunity 10 5 457--69
Complement receptor 2 polymorphisms associated with systemic lupus erythematosus modulate alternative splicing.
Genetic factors influence susceptibility to systemic lupus erythematosus (SLE). A recent family-based analysis in Caucasian and Chinese populations provided evidence for association of single-nucleotide polymorphisms (SNPs) in the complement receptor 2 (CR2/CD21) gene with SLE. Here we confirmed this result in a case-control analysis of an independent European-derived population including 2084 patients with SLE and 2853 healthy controls. A haplotype formed by the minor alleles of three CR2 SNPs (rs1048971,rs17615,rs4308977) showed significant association with decreased risk of SLE (30.4% in cases vs 32.6% in controls,P=0.016,OR=0.90 (0.82-0.98)). Two of these SNPs are in exon 10,directly 5' of an alternatively spliced exon preferentially expressed in follicular dendritic cells (FDC),and the third is in the alternatively spliced exon. Effects of these SNPs and a fourth SNP in exon 11 (rs17616) on alternative splicing were evaluated. We found that the minor alleles of these SNPs decreased splicing efficiency of exon 11 both in vitro and ex vivo. These findings further implicate CR2 in the pathogenesis of SLE and suggest that CR2 variants alter the maintenance of tolerance and autoantibody production in the secondary lymphoid tissues where B cells and FDCs interact.
View Publication
产品号#:
19054
19054RF
产品名:
EasySep™人B细胞富集试剂盒
RoboSep™ 人B细胞富集试剂盒含滤芯吸头
Tan Q et al. (JAN 2018)
JCI insight 3 1
Activation-induced cytidine deaminase deficiency accelerates autoimmune diabetes in NOD mice.
B cells play an important role in type 1 diabetes (T1D) development. However,the role of B cell activation-induced cytidine deaminase (AID) in diabetes development is not clear. We hypothesized that AID is important in the immunopathogenesis of T1D. To test this hypothesis,we generated AID-deficient (AID-/-) NOD mice. We found that AID-/-NOD mice developed accelerated T1D,with worse insulitis and high levels of anti-insulin autoantibody in the circulation. Interestingly,neither maternal IgG transferred through placenta,nor IgA transferred through milk affected the accelerated diabetes development. AID-/-NOD mice showed increased activation and proliferation of B and T cells. We found enhanced T-B cell interactions in AID-/-NOD mice,with increased T-bet and IFN-γ expression in CD4+ T cells in the presence of AID-/- B cells. Moreover,excessive lymphoid expansion was observed in AID-/-NOD mice. Importantly,antigen-specific BDC2.5 CD4+ T cells caused more rapid onset of diabetes when cotransferred with AID-/- B cells than when cotransferred with AID+/+ B cells. Thus,our study provides insights into the role of AID in T1D. Our data also suggest that AID is a negative regulator of immune tolerance and ablation of AID can lead to exacerbated islet autoimmunity and accelerated T1D development.
View Publication
产品号#:
19854
19854RF
产品名:
EasySep™小鼠B细胞分选试剂盒
RoboSep™ 小鼠B细胞分选试剂盒
Le Y et al. (MAR 2005)
Journal of immunology (Baltimore,Md. : 1950) 174 5 2582--90
CXC chemokine ligand 12-induced focal adhesion kinase activation and segregation into membrane domains is modulated by regulator of G protein signaling 1 in pro-B cells.
CXCL12-induced chemotaxis and adhesion to VCAM-1 decrease as B cells differentiate in the bone marrow. However,the mechanisms that regulate CXCL12/CXCR4-mediated signaling are poorly understood. We report that after CXCL12 stimulation of progenitor B cells,focal adhesion kinase (FAK) and PI3K are inducibly recruited to raft-associated membrane domains. After CXCL12 stimulation,phosphorylated FAK is also localized in membrane domains. The CXCL12/CXCR4-FAK pathway is membrane cholesterol dependent and impaired by metabolic inhibitors of G(i),Src family,and the GTPase-activating protein,regulator of G protein signaling 1 (RGS1). In the bone marrow,RGS1 mRNA expression is low in progenitor B cells and high in mature B cells,implying developmental regulation of CXCL12/CXCR4 signaling by RGS1. CXCL12-induced chemotaxis and adhesion are impaired when FAK recruitment and phosphorylation are inhibited by either membrane cholesterol depletion or overexpression of RGS1 in progenitor B cells. We conclude that the recruitment of signaling molecules to specific membrane domains plays an important role in CXCL12/CXCR4-induced cellular responses.
View Publication
产品号#:
产品名:
Balakrishnan K et al. (OCT 2006)
Blood 108 7 2392--8
Forodesine, an inhibitor of purine nucleoside phosphorylase, induces apoptosis in chronic lymphocytic leukemia cells.
Purine nucleoside phosphorylase (PNP) deficiency in humans results in T lymphocytopenia. Forodesine,a potent inhibitor of PNP,was designed based on the transition-state structure stabilized by the enzyme. Previous studies established that forodesine in the presence of deoxyguanosine (dGuo) inhibits the proliferation of T lymphocytes. A phase 1 clinical trial of forodesine in T-cell malignancies demonstrated significant antileukemic activity with an increase in intracellular dGuo triphosphate (dGTP). High accumulation of dGTP in T cells may be dependent on the levels of deoxynucleoside kinases. Because B-cell chronic lymphocytic leukemia (B-CLL) cells have high activity of deoxycytidine kinase (dCK),we hypothesized that these lymphocytes would respond to forodesine. This postulate was tested in primary lymphocytes during in vitro investigations. Lymphocytes from 12 patients with CLL were incubated with forodesine and dGuo. These CLL cells showed a wide variation in the accumulation of intracellular dGTP without any effect on other deoxynucleotides. This was associated with DNA damage-induced p53 stabilization,phosphorylation of p53 at Ser15,and activation of p21. The dGTP accumulation was related to induction of apoptosis measured by caspase activation,changes in mitochondrial membrane potential,and PARP cleavage. Based on these data,a phase 2 clinical trial of forodesine has been initiated for CLL patients.
View Publication
产品号#:
19051
19051RF
19054
19054RF
产品名:
EasySep™人T细胞富集试剂盒
RoboSep™ 人T细胞富集试剂盒含滤芯吸头
EasySep™人B细胞富集试剂盒
RoboSep™ 人B细胞富集试剂盒含滤芯吸头
Finstad SL et al. (JUL 2007)
Journal of virology 81 13 7274--9
Diminished potential for B-lymphoid differentiation after murine leukemia virus infection in vivo and in EML hematopoietic progenitor cells.
Infection with a recombinant murine-feline gammaretrovirus,MoFe2,or with the parent virus,Moloney murine leukemia virus,caused significant reduction in B-lymphoid differentiation of bone marrow at 2 to 8 weeks postinfection. The suppression was selective,in that myeloid potential was significantly increased by infection. Analysis of cell surface markers and immunoglobulin H gene rearrangements in an in vitro model demonstrated normal B-lymphoid differentiation after infection but significantly reduced viability of differentiating cells. This reduction in viability may confer a selective advantage on undifferentiated lymphoid progenitors in the bone marrow of gammaretrovirus-infected animals and thereby contribute to the establishment of a premalignant state.
View Publication
产品号#:
03630
03434
03444
产品名:
MethoCult™ M3630
MethoCult™ GF M3434
MethoCult™ GF M3434
Nguyen CQ et al. (JUL 2007)
Journal of immunology (Baltimore,Md. : 1950) 179 1 382--90
IL-4-STAT6 signal transduction-dependent induction of the clinical phase of Sjögren's syndrome-like disease of the nonobese diabetic mouse.
NOD.B10-H2(b) and NOD/LtJ mice manifest,respectively,many features of primary and secondary Sjögren's syndrome (SjS),an autoimmune disease affecting primarily the salivary and lacrimal glands leading to xerostomia (dry mouth) and xerophthalmia (dry eyes). B lymphocytes play a central role in the onset of SjS with clinical manifestations dependent on the appearance of autoantibodies reactive to multiple components of acinar cells. Previous studies with NOD.IL4(-/-) and NOD.B10-H2(b).IL4(-/-) mice suggest that the Th2 cytokine,IL-4,plays a vital role in the development and onset of SjS-like disease in the NOD mouse model. To investigate the molecular mechanisms by which IL-4 controls SjS development,a Stat6 gene knockout mouse,NOD.B10-H2(b).C-Stat6(-/-),was constructed and its disease profile was defined and compared with that of NOD.B10-H2(b).C-Stat6(+/+) mice. As the NOD.B10-H2(b).C-Stat6(-/-) mice aged from 4 to 24 wk,they exhibited leukocyte infiltration of the exocrine glands,produced anti-nuclear autoantibodies,and showed loss and gain of saliva-associated proteolytic enzymes,similar to NOD.B10-H2(b).C-Stat6(+/+) mice. In contrast,NOD.B10-H2(b).C-Stat6(-/-) mice failed to develop glandular dysfunction,maintaining normal saliva flow rates. NOD.B10-H2(b).C-Stat6(-/-) mice were found to lack IgG1 isotype-specific anti-muscarinic acetylcholine type-3 receptor autoantibodies. Furthermore,the IgG fractions from NOD.B10-H2(b).C-Stat6(-/-) sera were unable to induce glandular dysfunction when injected into naive recipient C57BL/6 mice. NOD.B10-H2(b).C-Stat6(-/-) mice,like NOD.B10-H2(b).IL4(-/-) mice,are unable to synthesize IgG1 Abs,an observation that correlates with an inability to develop end-stage clinical SjS-like disease. These data imply a requirement for the IL-4/STAT6-pathway for onset of the clinical phase of SjS-like disease in the NOD mouse model.
View Publication
产品号#:
18754
18754RF
产品名:
挂图
Human Immune Cytokines
Infographic of key cytokines for expansion, differentiation and characterization of major immune cell types
Nova-Lamperti E et al. (JAN 2016)
Scientific Reports 6 20044
IL-10-produced by human transitional B-cells down-regulates CD86 expression on B-cells leading to inhibition of CD4+T-cell responses.
A novel subset of human regulatory B-cells has recently been described. They arise from within the transitional B-cell subpopulation and are characterised by the production of IL-10. They appear to be of significant importance in regulating T-cell immunity in vivo. Despite this important function,the molecular mechanisms by which they control T-cell activation are incompletely defined. Here we show that transitional B-cells produced more IL-10 and expressed higher levels of IL-10 receptor after CD40 engagement compared to other B-cell subsets. Furthermore,under this stimulatory condition,CD86 expressed by transitional B-cells was down regulated and T-cell proliferation was reduced. We provide evidence to demonstrate that the down-regulation of CD86 expression by transitional B-cells was due to the autocrine effect of IL-10,which in turn leads to decreased T-cell proliferation and TNF-α production. This analysis was further extended to peripheral B-cells in kidney transplant recipients. We observed that B-cells from patients tolerant to the graft maintained higher IL-10 production after CD40 ligation,which correlates with lower CD86 expression compared to patients with chronic rejection. Hence,the results obtained in this study shed light on a new alternative mechanism by which transitional B-cells inhibit T-cell proliferation and cytokine production.
View Publication
产品号#:
15022
15062
15024
15064
产品名:
RosetteSep™人CD4+ T细胞富集抗体混合物
RosetteSep™人CD4+ T细胞富集抗体混合物
RosetteSep™人B细胞富集抗体混合物
RosetteSep™人B细胞富集抗体混合物
Valsecchi R et al. (APR 2016)
Blood 127 16 1987--97
HIF-1α regulates the interaction of chronic lymphocytic leukemia cells with the tumor microenvironment.
Hypoxia-inducible transcription factors (HIFs) regulate a wide array of adaptive responses to hypoxia and are often activated in solid tumors and hematologic malignancies due to intratumoral hypoxia and emerging new layers of regulation. We found that in chronic lymphocytic leukemia (CLL),HIF-1α is a novel regulator of the interaction of CLL cells with protective leukemia microenvironments and,in turn,is regulated by this interaction in a positive feedback loop that promotes leukemia survival and propagation. Through unbiased microarray analysis,we found that in CLL cells,HIF-1α regulates the expression of important chemokine receptors and cell adhesion molecules that control the interaction of leukemic cells with bone marrow and spleen microenvironments. Inactivation of HIF-1α impairs chemotaxis and cell adhesion to stroma,reduces bone marrow and spleen colonization in xenograft and allograft CLL mouse models,and prolongs survival in mice. Of interest,we found that in CLL cells,HIF-1α is transcriptionally regulated after coculture with stromal cells. Furthermore,HIF-1α messenger RNA levels vary significantly within CLL patients and correlate with the expression of HIF-1α target genes,including CXCR4,thus further emphasizing the relevance of HIF-1α expression to CLL pathogenesis.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




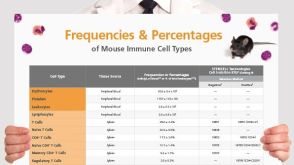 挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice
挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice 挂图Human Immune Cytokines Infographic of key cytokines for expansion, differentiation and characterization of major immune cell types
挂图Human Immune Cytokines Infographic of key cytokines for expansion, differentiation and characterization of major immune cell types

 沪公网安备31010102008431号
沪公网安备31010102008431号