A critical challenge to deciphering the pathophysiology of neurodevelopmental disease is identifying which of the myriad abnormalities that emerge during CNS maturation persist to contribute to long-term brain dysfunction. Childhood-onset dystonia caused by a loss-of-function mutation in the AAA+ protein torsinA exemplifies this challenge. Neurons lacking torsinA develop transient nuclear envelope (NE) malformations during CNS maturation,but no NE defects are described in mature torsinA null neurons. We find that during postnatal CNS maturation torsinA null neurons develop mislocalized and dysfunctional nuclear pore complexes (NPC) that lack NUP358,normally added late in NPC biogenesis. SUN1,a torsinA-related molecule implicated in interphase NPC biogenesis,also exhibits localization abnormalities. Whereas SUN1 and associated nuclear membrane abnormalities resolve in juvenile mice,NPC defects persist into adulthood. These findings support a role for torsinA function in NPC biogenesis during neuronal maturation and implicate altered NPC function in dystonia pathophysiology.
View Publication
产品号#:
05711
05790
05792
05793
05794
05795
100-1281
产品名:
NeuroCult™ SM1 神经添加物
BrainPhys™神经元培养基
BrainPhys™神经元培养基和SM1试剂盒
BrainPhys™ 神经元培养基N2-A和SM1试剂盒
BrainPhys™原代神经元试剂盒
BrainPhys™ hPSC 神经元试剂盒
NeuroCult™ SM1 神经添加物
Paquet D et al. (MAY 2016)
Nature 533 7601 125--129
Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9
The bacterial CRISPR/Cas9 system allows sequence-specific gene editing in many organisms and holds promise as a tool to generate models of human diseases,for example,in human pluripotent stem cells. CRISPR/Cas9 introduces targeted double-stranded breaks (DSBs) with high efficiency,which are typically repaired by non-homologous end-joining (NHEJ) resulting in nonspecific insertions,deletions or other mutations (indels). DSBs may also be repaired by homology-directed repair (HDR) using a DNA repair template,such as an introduced single-stranded oligo DNA nucleotide (ssODN),allowing knock-in of specific mutations. Although CRISPR/Cas9 is used extensively to engineer gene knockouts through NHEJ,editing by HDR remains inefficient and can be corrupted by additional indels,preventing its widespread use for modelling genetic disorders through introducing disease-associated mutations. Furthermore,targeted mutational knock-in at single alleles to model diseases caused by heterozygous mutations has not been reported. Here we describe a CRISPR/Cas9-based genome-editing framework that allows selective introduction of mono- and bi-allelic sequence changes with high efficiency and accuracy. We show that HDR accuracy is increased dramatically by incorporating silent CRISPR/Cas-blocking mutations along with pathogenic mutations,and establish a method termed 'CORRECT' for scarless genome editing. By characterizing and exploiting a stereotyped inverse relationship between a mutation's incorporation rate and its distance to the DSB,we achieve predictable control of zygosity. Homozygous introduction requires a guide RNA targeting close to the intended mutation,whereas heterozygous introduction can be accomplished by distance-dependent suboptimal mutation incorporation or by use of mixed repair templates. Using this approach,we generated human induced pluripotent stem cells with heterozygous and homozygous dominant early onset Alzheimer's disease-causing mutations in amyloid precursor protein (APP(Swe)) and presenilin 1 (PSEN1(M146V)) and derived cortical neurons,which displayed genotype-dependent disease-associated phenotypes. Our findings enable efficient introduction of specific sequence changes with CRISPR/Cas9,facilitating study of human disease.
View Publication
B. S. Souza et al. (dec 2016)
Scientific Reports 6 1 39775
Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells
Zika virus (ZIKV) infection has been associated with severe complications both in the developing and adult nervous system. To investigate the deleterious effects of ZIKV infection,we used human neural progenitor cells (NPC),derived from induced pluripotent stem cells (iPSC). We found that NPC are highly susceptible to ZIKV and the infection results in cell death. ZIKV infection led to a marked reduction in cell proliferation,ultrastructural alterations and induction of autophagy. Induction of apoptosis of Sox2 + cells was demonstrated by activation of caspases 3/7,8 and 9,and by ultrastructural and flow cytometry analyses. ZIKV-induced death of Sox2 + cells was prevented by incubation with the pan-caspase inhibitor,Z-VAD-FMK. By confocal microscopy analysis we found an increased number of cells with supernumerary centrosomes. Live imaging showed a significant increase in mitosis abnormalities,including multipolar spindle,chromosome laggards,micronuclei and death of progeny after cell division. FISH analysis for chromosomes 12 and 17 showed increased frequency of aneuploidy,such as monosomy,trisomy and polyploidy. Our study reinforces the link between ZIKV and abnormalities in the developing human brain,including microcephaly.
View Publication
产品号#:
05832
05833
19851
19851RF
19852
19852RF
19854
19854RF
05835
05839
产品名:
STEMdiff™ 神经花环选择试剂
STEMdiff™神经前体细胞培养基
EasySep™小鼠T细胞分选试剂盒
RoboSep™ 小鼠T细胞分选试剂盒
EasySep™小鼠CD4+ T细胞分选试剂盒
RoboSep™ 小鼠CD4+ T细胞分选试剂盒
EasySep™小鼠B细胞分选试剂盒
RoboSep™ 小鼠B细胞分选试剂盒
STEMdiff™ 神经诱导培养基
STEMdiff™ 神经诱导培养基
Tagliafierro L et al. (NOV 2017)
Alzheimer's & dementia : the journal of the Alzheimer's Association 13 11 1237--1250
Genetic analysis of α-synuclein 3' untranslated region and its corresponding microRNAs in relation to Parkinson's disease compared to dementia with Lewy bodies.
INTRODUCTION The α-synuclein (SNCA) gene has been implicated in the etiology of Parkinson's disease (PD) and dementia with Lewy bodies (DLB). METHODS A computational analysis of SNCA 3' untranslated region to identify potential microRNA (miRNA) binding sites and quantitative real-time polymerase chain reaction (PCR) to determine their expression in isogenic induced pluripotent stem cell-derived dopaminergic and cholinergic neurons as a model of PD and DLB,respectively,were performed. In addition,we performed a deep sequencing analysis of the SNCA 3' untranslated region of autopsy-confirmed cases of PD,DLB,and normal controls,followed by genetic association analysis of the identified variants. RESULTS We identified four miRNA binding sites and observed a neuronal-type-specific expression profile for each miRNA in the different isogenic induced pluripotent stem cell-derived dopaminergic and cholinergic neurons. Furthermore,we found that the short structural variant rs777296100-polyT was moderately associated with DLB but not with PD. DISCUSSION We suggest that the regulation of SNCA expression through miRNAs is neuronal-type-specific and possibly plays a part in the phenotypic heterogeneity of synucleinopathies. Furthermore,genetic variability in the SNCA gene may contribute to synucleinopathies in a pathology-specific manner.
View Publication
产品号#:
05790
05792
05793
05794
05795
产品名:
BrainPhys™神经元培养基
BrainPhys™神经元培养基和SM1试剂盒
BrainPhys™ 神经元培养基N2-A和SM1试剂盒
BrainPhys™原代神经元试剂盒
BrainPhys™ hPSC 神经元试剂盒
Deng M et al. (JAN 2018)
European Journal of Neuroscience 47 2 150--157
Preservation of neuronal functions by exosomes derived from different human neural cell types under ischemic conditions
Stem cell-based therapies have been reported in protecting cerebral infarction-induced neuronal dysfunction and death. However,most studies used rat/mouse neuron as model cell when treated with stem cell or exosomes. Whether these findings can be translated from rodent to humans has been in doubt. Here,we used human embryonic stem cell-derived neurons to detect the protective potential of exosomes against ischemia. Neurons were treated with in vitro oxygen-glucose deprivation (OGD) for 1 h. For treatment group,different exosomes were derived from neuron,embryonic stem cell,neural progenitor cell and astrocyte differentiated from H9 human embryonic stem cell and added to culture medium 30 min after OGD (100 μg/mL). Western blotting was performed 12 h after OGD,while cell counting and electrophysiological recording were performed 48 h after OGD. We found that these exosomes attenuated OGD-induced neuronal death,Mammalian target of rapamycin (mTOR),pro-inflammatory and apoptotic signaling pathway changes,as well as basal spontaneous synaptic transmission inhibition in varying degrees. The results implicate the protective effect of exosomes on OGD-induced neuronal death and dysfunction in human embryonic stem cell-derived neurons,potentially through their modulation on mTOR,pro-inflammatory and apoptotic signaling pathways.
View Publication
产品号#:
05711
05790
05792
05793
05794
05795
100-1281
产品名:
NeuroCult™ SM1 神经添加物
BrainPhys™神经元培养基
BrainPhys™神经元培养基和SM1试剂盒
BrainPhys™ 神经元培养基N2-A和SM1试剂盒
BrainPhys™原代神经元试剂盒
BrainPhys™ hPSC 神经元试剂盒
NeuroCult™ SM1 神经添加物
Xia G et al. (JUN 2015)
Stem cells (Dayton,Ohio) 33 6 1829--38
Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 induced pluripotent stem cell-derived neural stem cells.
Myotonic dystrophy type 1 (DM1) is caused by expanded CTG repeats in the 3'-untranslated region (3' UTR) of the DMPK gene. Correcting the mutation in DM1 stem cells would be an important step toward autologous stem cell therapy. The objective of this study is to demonstrate in vitro genome editing to prevent production of toxic mutant transcripts and reverse phenotypes in DM1 stem cells. Genome editing was performed in DM1 neural stem cells (NSCs) derived from human DM1 induced pluripotent stem (iPS) cells. An editing cassette containing SV40/bGH polyA signals was integrated upstream of the CTG repeats by TALEN-mediated homologous recombination (HR). The expression of mutant CUG repeats transcript was monitored by nuclear RNA foci,the molecular hallmarks of DM1,using RNA fluorescence in situ hybridization. Alternative splicing of microtubule-associated protein tau (MAPT) and muscleblind-like (MBNL) proteins were analyzed to further monitor the phenotype reversal after genome modification. The cassette was successfully inserted into DMPK intron 9 and this genomic modification led to complete disappearance of nuclear RNA foci. MAPT and MBNL 1,2 aberrant splicing in DM1 NSCs were reversed to normal pattern in genome-modified NSCs. Genome modification by integration of exogenous polyA signals upstream of the DMPK CTG repeat expansion prevents the production of toxic RNA and leads to phenotype reversal in human DM1 iPS-cells derived stem cells. Our data provide proof-of-principle evidence that genome modification may be used to generate genetically modified progenitor cells as a first step toward autologous cell transfer therapy for DM1.
View Publication
A homozygous loss-of-function CAMK2A mutation causes growth delay, frequent seizures and severe intellectual disability.
Calcium/calmodulin-dependent protein kinase II (CAMK2) plays fundamental roles in synaptic plasticity that underlies learning and memory. Here,we describe a new recessive neurodevelopmental syndrome with global developmental delay,seizures and intellectual disability. Using linkage analysis and exome sequencing,we found that this disease maps to chromosome 5q31.1-q34 and is caused by a biallelic germline mutation in CAMK2A. The missense mutation,p.His477Tyr is located in the CAMK2A association domain that is critical for its function and localization. Biochemically,the p.His477Tyr mutant is defective in self-oligomerization and unable to assemble into the multimeric holoenzyme.In vivo,CAMK2AH477Y failed to rescue neuronal defects in C. elegans lacking unc-43,the ortholog of human CAMK2A. In vitro,neurons derived from patient iPSCs displayed profound synaptic defects. Together,our data demonstrate that a recessive germline mutation in CAMK2A leads to neurodevelopmental defects in humans and suggest that dysfunctional CAMK2 paralogs may contribute to other neurological disorders.
View Publication
产品号#:
05790
05792
05793
05794
05795
85850
85857
85870
85875
产品名:
BrainPhys™神经元培养基
BrainPhys™神经元培养基和SM1试剂盒
BrainPhys™ 神经元培养基N2-A和SM1试剂盒
BrainPhys™原代神经元试剂盒
BrainPhys™ hPSC 神经元试剂盒
mTeSR™1
mTeSR™1
S. Bell et al. (JUL 2018)
Stem cell reports 11 1 183--196
Disruption of GRIN2B Impairs Differentiation in Human Neurons.
Heterozygous loss-of-function mutations in GRIN2B,a subunit of the NMDA receptor,cause intellectual disability and language impairment. We developed clonal models of GRIN2B deletion and loss-of-function mutations in a region coding for the glutamate binding domain in human cells and generated neurons from a patient harboring a missense mutation in the same domain. Transcriptome analysis revealed extensive increases in genes associated with cell proliferation and decreases in genes associated with neuron differentiation,a result supported by extensive protein analyses. Using electrophysiology and calcium imaging,we demonstrate that NMDA receptors are present on neural progenitor cells and that human mutations in GRIN2B can impair calcium influx and membrane depolarization even in a presumed undifferentiated cell state,highlighting an important role for non-synaptic NMDA receptors. It may be this function,in part,which underlies the neurological disease observed in patients with GRIN2B mutations.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




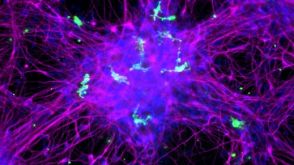 实验方案How to Co-Culture Human Pluripotent Stem Cell (hPSC)-Derived Forebrain Neurons and Microglia
实验方案How to Co-Culture Human Pluripotent Stem Cell (hPSC)-Derived Forebrain Neurons and Microglia


 沪公网安备31010102008431号
沪公网安备31010102008431号