Martí et al. (APR 2016)
Molecular Neurobiology 53 5 2857--2868
RTP801 Is Involved in Mutant Huntingtin-Induced Cell Death
RTP801 expression is induced by cellular stress and has a pro-apoptotic function in non-proliferating differentiated cells such as neurons. In several neurodegenerative disorders,including Parkinson's disease and Alzheimer's disease,elevated levels of RTP801 have been observed,which suggests a role for RTP801 in neuronal death. Neuronal death is also a pathological hallmark in Huntington's disease (HD),an inherited neurodegenerative disorder caused by a CAG repeat expansion in the huntingtin gene. Currently,the exact mechanisms underlying mutant huntingtin (mhtt)-induced toxicity are still unclear. Here,we investigated whether RTP801 is involved in (mhtt)-induced cell death. Ectopic exon-1 mhtt elevated RTP801 mRNA and protein levels in nerve growth factor (NGF)-differentiated PC12 cells and in rat primary cortical neurons. In neuronal PC12 cells,mhtt also contributed to RTP801 protein elevation by reducing its proteasomal degradation rate,in addition to promoting RTP801 gene expression. Interestingly,silencing RTP801 expression with short hairpin RNAs (shRNAs) blocked mhtt-induced cell death in NGF-differentiated PC12 cells. However,RTP801 protein levels were not altered in the striatum of Hdh(Q7/Q111) and R6/1 mice,two HD models that display motor deficits but not neuronal death. Importantly,RTP801 protein levels were elevated in both neural telencephalic progenitors differentiated from HD patient-derived induced pluripotent stem cells and in the putamen and cerebellum of human HD postmortem brains. Taken together,our results suggest that RTP801 is a novel downstream effector of mhtt-induced toxicity and that it may be relevant to the human disease.
View Publication
产品号#:
05850
05857
05870
05875
85850
85857
85870
85875
产品名:
mTeSR™1
mTeSR™1
S. Bell et al. (JUL 2018)
Stem cell reports 11 1 183--196
Disruption of GRIN2B Impairs Differentiation in Human Neurons.
Heterozygous loss-of-function mutations in GRIN2B,a subunit of the NMDA receptor,cause intellectual disability and language impairment. We developed clonal models of GRIN2B deletion and loss-of-function mutations in a region coding for the glutamate binding domain in human cells and generated neurons from a patient harboring a missense mutation in the same domain. Transcriptome analysis revealed extensive increases in genes associated with cell proliferation and decreases in genes associated with neuron differentiation,a result supported by extensive protein analyses. Using electrophysiology and calcium imaging,we demonstrate that NMDA receptors are present on neural progenitor cells and that human mutations in GRIN2B can impair calcium influx and membrane depolarization even in a presumed undifferentiated cell state,highlighting an important role for non-synaptic NMDA receptors. It may be this function,in part,which underlies the neurological disease observed in patients with GRIN2B mutations.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒





 27:19
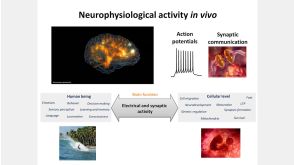
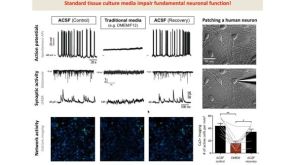
线上讲座BrainPhys™ Medium Supports the Physiological Activity of Neuronal Tissue in vitro发布日期: 07/22/2016
27:19
线上讲座BrainPhys™ Medium Supports the Physiological Activity of Neuronal Tissue in vitro发布日期: 07/22/2016 科学海报An In Vitro Assay to Determine the Neurotoxic Effects of Pharmacological Compounds
科学海报An In Vitro Assay to Determine the Neurotoxic Effects of Pharmacological Compounds


 沪公网安备31010102008431号
沪公网安备31010102008431号