Balakrishnan K et al. (OCT 2006)
Blood 108 7 2392--8
Forodesine, an inhibitor of purine nucleoside phosphorylase, induces apoptosis in chronic lymphocytic leukemia cells.
Purine nucleoside phosphorylase (PNP) deficiency in humans results in T lymphocytopenia. Forodesine,a potent inhibitor of PNP,was designed based on the transition-state structure stabilized by the enzyme. Previous studies established that forodesine in the presence of deoxyguanosine (dGuo) inhibits the proliferation of T lymphocytes. A phase 1 clinical trial of forodesine in T-cell malignancies demonstrated significant antileukemic activity with an increase in intracellular dGuo triphosphate (dGTP). High accumulation of dGTP in T cells may be dependent on the levels of deoxynucleoside kinases. Because B-cell chronic lymphocytic leukemia (B-CLL) cells have high activity of deoxycytidine kinase (dCK),we hypothesized that these lymphocytes would respond to forodesine. This postulate was tested in primary lymphocytes during in vitro investigations. Lymphocytes from 12 patients with CLL were incubated with forodesine and dGuo. These CLL cells showed a wide variation in the accumulation of intracellular dGTP without any effect on other deoxynucleotides. This was associated with DNA damage-induced p53 stabilization,phosphorylation of p53 at Ser15,and activation of p21. The dGTP accumulation was related to induction of apoptosis measured by caspase activation,changes in mitochondrial membrane potential,and PARP cleavage. Based on these data,a phase 2 clinical trial of forodesine has been initiated for CLL patients.
View Publication
产品号#:
19051
19051RF
19054
19054RF
产品名:
EasySep™人T细胞富集试剂盒
RoboSep™ 人T细胞富集试剂盒含滤芯吸头
EasySep™人B细胞富集试剂盒
RoboSep™ 人B细胞富集试剂盒含滤芯吸头
Le Y et al. (MAR 2005)
Journal of immunology (Baltimore,Md. : 1950) 174 5 2582--90
CXC chemokine ligand 12-induced focal adhesion kinase activation and segregation into membrane domains is modulated by regulator of G protein signaling 1 in pro-B cells.
CXCL12-induced chemotaxis and adhesion to VCAM-1 decrease as B cells differentiate in the bone marrow. However,the mechanisms that regulate CXCL12/CXCR4-mediated signaling are poorly understood. We report that after CXCL12 stimulation of progenitor B cells,focal adhesion kinase (FAK) and PI3K are inducibly recruited to raft-associated membrane domains. After CXCL12 stimulation,phosphorylated FAK is also localized in membrane domains. The CXCL12/CXCR4-FAK pathway is membrane cholesterol dependent and impaired by metabolic inhibitors of G(i),Src family,and the GTPase-activating protein,regulator of G protein signaling 1 (RGS1). In the bone marrow,RGS1 mRNA expression is low in progenitor B cells and high in mature B cells,implying developmental regulation of CXCL12/CXCR4 signaling by RGS1. CXCL12-induced chemotaxis and adhesion are impaired when FAK recruitment and phosphorylation are inhibited by either membrane cholesterol depletion or overexpression of RGS1 in progenitor B cells. We conclude that the recruitment of signaling molecules to specific membrane domains plays an important role in CXCL12/CXCR4-induced cellular responses.
View Publication
产品号#:
产品名:
Nguyen CQ et al. (JUL 2007)
Journal of immunology (Baltimore,Md. : 1950) 179 1 382--90
IL-4-STAT6 signal transduction-dependent induction of the clinical phase of Sjögren's syndrome-like disease of the nonobese diabetic mouse.
NOD.B10-H2(b) and NOD/LtJ mice manifest,respectively,many features of primary and secondary Sjögren's syndrome (SjS),an autoimmune disease affecting primarily the salivary and lacrimal glands leading to xerostomia (dry mouth) and xerophthalmia (dry eyes). B lymphocytes play a central role in the onset of SjS with clinical manifestations dependent on the appearance of autoantibodies reactive to multiple components of acinar cells. Previous studies with NOD.IL4(-/-) and NOD.B10-H2(b).IL4(-/-) mice suggest that the Th2 cytokine,IL-4,plays a vital role in the development and onset of SjS-like disease in the NOD mouse model. To investigate the molecular mechanisms by which IL-4 controls SjS development,a Stat6 gene knockout mouse,NOD.B10-H2(b).C-Stat6(-/-),was constructed and its disease profile was defined and compared with that of NOD.B10-H2(b).C-Stat6(+/+) mice. As the NOD.B10-H2(b).C-Stat6(-/-) mice aged from 4 to 24 wk,they exhibited leukocyte infiltration of the exocrine glands,produced anti-nuclear autoantibodies,and showed loss and gain of saliva-associated proteolytic enzymes,similar to NOD.B10-H2(b).C-Stat6(+/+) mice. In contrast,NOD.B10-H2(b).C-Stat6(-/-) mice failed to develop glandular dysfunction,maintaining normal saliva flow rates. NOD.B10-H2(b).C-Stat6(-/-) mice were found to lack IgG1 isotype-specific anti-muscarinic acetylcholine type-3 receptor autoantibodies. Furthermore,the IgG fractions from NOD.B10-H2(b).C-Stat6(-/-) sera were unable to induce glandular dysfunction when injected into naive recipient C57BL/6 mice. NOD.B10-H2(b).C-Stat6(-/-) mice,like NOD.B10-H2(b).IL4(-/-) mice,are unable to synthesize IgG1 Abs,an observation that correlates with an inability to develop end-stage clinical SjS-like disease. These data imply a requirement for the IL-4/STAT6-pathway for onset of the clinical phase of SjS-like disease in the NOD mouse model.
View Publication
产品号#:
18754
18754RF
产品名:
Griffin DO et al. (JAN 2011)
The Journal of experimental medicine 208 1 67--80
Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70-.
B1 cells differ in many ways from conventional B cells,most prominently in the production of natural immunoglobulin,which is vitally important for protection against pathogens. B1 cells have also been implicated in the pathogenesis of autoimmune dyscrasias and malignant diseases. It has been impossible to accurately study B1 cells during health and illness because the nature of human B1 cells has not been successfully defined. This has produced controversy regarding the existence of human B1 cells. Here,we determined the phenotype of human B1 cells by testing sort-purified B cell fractions for three fundamental B1 cell functions based on mouse studies: spontaneous IgM secretion,efficient T cell stimulation,and tonic intracellular signaling. We found that a small population of CD20(+)CD27(+)CD43(+) cells present in both umbilical cord and adult peripheral blood fulfilled these criteria and expressed a skewed B cell receptor repertoire. These B cells express little or no surface CD69 and CD70,both of which are markedly up-regulated after activation of CD20(+)CD27(-)CD43(-) (naive) and CD20(+)CD27(+)CD43(-) (memory) B cells. This work identifies human B1 cells as CD20(+)CD27(+)CD43(+)CD70(-). We determined that the proportion of B1 cells declines with age,which may contribute to disease susceptibility. Identification of human B1 cells provides a foundation for future studies on the nature and role of these cells in human disease.
View Publication
TEL-AML1 promotes development of specific hematopoietic lineages consistent with preleukemic activity.
The t(12;21)(p13;q22) translocation is the most common chromosomal abnormality yet identified in any pediatric leukemia and gives rise to the TEL-AML1 fusion product. To investigate the effects of TEL-AML1 on hematopoiesis,fetal liver hematopoietic progenitor cells (HPCs) were transduced with retroviral vectors expressing this fusion protein. We show that TEL-AML1 dramatically alters differentiation of HPCs in vitro,preferentially promoting B-lymphocyte development,enhancing self-renewal of B-cell precursors,and leading to the establishment of long-term growth factor-dependent pre-B-cell lines. However,it had no effect on myeloid development in vitro. Further experiments were performed to determine whether TEL-AML1 also demonstrates lineage-specific activity in vivo. TEL-AML1-expressing HPCs displayed a competitive advantage in reconstituting both B-cell and myeloid lineages in vivo but had no effect on reconstitution of the T-cell lineage. Despite promoting these alterations in hematopoiesis,TEL-AML1 did not induce leukemia in transplanted mice. Our study provides a unique insight into the role of TEL-AML1 in leukemia predisposition and a potential model to study the mechanism of leukemogenesis associated with this fusion.
View Publication
产品号#:
03534
03231
产品名:
MethoCult™ GF M3534
MethoCult™ M3231
Coletta PL et al. (FEB 2004)
Blood 103 3 1050--8
Lymphodepletion in the ApcMin/+ mouse model of intestinal tumorigenesis.
Germ line mutations in the Adenomatous polyposis coli tumor suppressor gene cause a hereditary form of intestinal tumorigenesis in both mice and man. Here we show that in Apc(Min/+) mice,which carry a heterozygous germ line mutation at codon 850 of Apc,there is progressive loss of immature and mature thymocytes from approximately 80 days of age with complete regression of the thymus by 120 days. In addition,Apc(Min/+) mice show parallel depletion of splenic natural killer (NK) cells,immature B cells,and B progenitor cells in bone marrow due to complete loss of interleukin 7 (IL-7)-dependent B-cell progenitors. Using bone marrow transplantation experiments into wild-type recipients,we have shown that the capacity of transplanted Apc(Min/+) bone marrow cells for T- and B-cell development appears normal. In contrast,although the Apc(Min/+) bone marrow microenvironment supported short-term reconstitution with wild-type bone marrow,Apc(Min/+) animals that received transplants subsequently underwent lymphodepletion. Fibroblast colony-forming unit (CFU-F) colony assays revealed a significant reduction in colony-forming mesenchymal progenitor cells in the bone marrow of Apc(Min/+) mice compared with wild-type animals prior to the onset of lymphodepletion. This suggests that an altered bone marrow microenvironment may account for the selective lymphocyte depletion observed in this model of familial adenomatous polyposis.
View Publication
Iwasaki-Arai J et al. (MAY 2003)
The Journal of experimental medicine 197 10 1311--22
Enforced granulocyte/macrophage colony-stimulating factor signals do not support lymphopoiesis, but instruct lymphoid to myelomonocytic lineage conversion.
We evaluated the effects of ectopic granulocyte/macrophage colony-stimulating factor (GM-CSF) signals on hematopoietic commitment and differentiation. Lineage-restricted progenitors purified from mice with the ubiquitous transgenic human GM-CSF receptor (hGM-CSFR) were used for the analysis. In cultures with hGM-CSF alone,hGM-CSFR-expressing (hGM-CSFR+) granulocyte/monocyte progenitors (GMPs) and megakaryocyte/erythrocyte progenitors (MEPs) exclusively gave rise to granulocyte/monocyte (GM) and megakaryocyte/erythroid (MegE) colonies,respectively,providing formal proof that GM-CSF signals support the GM and MegE lineage differentiation without affecting the physiological myeloid fate. hGM-CSFR transgenic mice were crossed with mice deficient in interleukin (IL)-7,an essential cytokine for T and B cell development. Administration of hGM-CSF in these mice could not restore T or B lymphopoiesis,indicating that enforced GM-CSF signals cannot substitute for IL-7 to promote lymphopoiesis. Strikingly,textgreater50% hGM-CSFR+ common lymphoid progenitors (CLPs) and textgreater20% hGM-CSFR+ pro-T cells gave rise to granulocyte,monocyte,and/or myeloid dendritic cells,but not MegE lineage cells in the presence of hGM-CSF. Injection of hGM-CSF into mice transplanted with hGM-CSFR+ CLPs blocked their lymphoid differentiation,but induced development of GM cells in vivo. Thus,hGM-CSF transduces permissive signals for myeloerythroid differentiation,whereas it transmits potent instructive signals for the GM differentiation to CLPs and early T cell progenitors. These data suggest that a majority of CLPs and a fraction of pro-T cells possess plasticity for myelomonocytic differentiation that can be activated by ectopic GM-CSF signals,supporting the hypothesis that the down-regulation of GM-CSFR is a critical event in producing cells with a lymphoid-restricted lineage potential.
View Publication
产品号#:
04100
产品名:
MethoCult™ H4100
Yates F et al. (DEC 2002)
Blood 100 12 3942--9
Gene therapy of RAG-2-/- mice: sustained correction of the immunodeficiency.
Patients with mutations of either RAG-1 or RAG-2 genes suffer from severe combined immunodeficiency (SCID) characterized by the lack of T and B lymphocytes. The only curative treatment today consists of hematopoietic stem cell (HSC) transplantation,which is only partially successful in the absence of an HLA genoidentical donor,thus justifying research to find an alternative therapeutic approach. To this end,RAG-2-deficient mice were used to test whether retrovirally mediated ex vivo gene transfer into HSCs could provide long-term correction of the immunologic deficiency. Murine RAG-2-/-Sca-1(+) selected bone marrow cells were transduced with a modified Moloney leukemia virus (MLV)-based MND (myeloproliferative sarcoma virus enhancer,negative control region deleted,dl587rev primer-binding site substituted) retroviral vector containing the RAG-2 cDNA and transplanted into RAG-2-/- sublethally irradiated mice (3Gy). Two months later,T- and B-cell development was achieved in all mice. Diverse repertoire of T cells as well as proliferative capacity in the presence of mitogens,allogeneic cells,and keyhole limpet hemocyanin (KLH) were shown. B-cell function as shown by serum Ig levels and antibody response to a challenge by KLH also developed. Lymphoid subsets and function were shown to be stable over a one-year period without evidence of any detectable toxicity. Noteworthy,a selective advantage for transduced lymphoid cells was evidenced by comparative provirus quantification in lymphoid and myeloid lineages. Altogether,this study demonstrates the efficiency of ex vivo RAG-2 gene transfer in HSCs to correct the immune deficiency of RAG-2-/- mice,constituting a significant step toward clinical application.
View Publication
PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice.
The tumor suppressor gene phosphatase and tensin homolog (PTEN) is inactivated in many human cancers. However,it is unknown whether PTEN functions as a tumor suppressor in human Philadelphia chromosome-positive leukemia that includes chronic myeloid leukemia (CML) and B-cell acute lymphoblastic leukemia (B-ALL) and is induced by the BCR-ABL oncogene. By using our mouse model of BCR-ABL-induced leukemias,we show that Pten is down-regulated by BCR-ABL in leukemia stem cells in CML and that PTEN deletion causes acceleration of CML development. In addition,overexpression of PTEN delays the development of CML and B-ALL and prolongs survival of leukemia mice. PTEN suppresses leukemia stem cells and induces cell-cycle arrest of leukemia cells. Moreover,PTEN suppresses B-ALL development through regulating its downstream gene Akt1. These results demonstrate a critical role of PTEN in BCR-ABL-induced leukemias and suggest a potential strategy for the treatment of Philadelphia chromosome-positive leukemia.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒






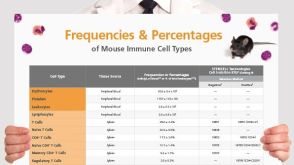 挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice
挂图Frequencies and Percentages of Mouse Immune Cell Types List of the frequencies of over 25 immune cell types in C57BL/6 mice
 沪公网安备31010102008431号
沪公网安备31010102008431号