
 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




技术资料
-

-
 27:19
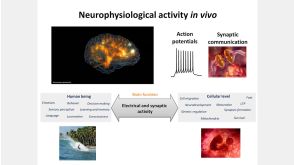
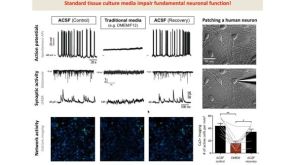
线上讲座BrainPhys™ Medium Supports the Physiological Activity of Neuronal Tissue in vitro发布日期: 07/22/2016
27:19
线上讲座BrainPhys™ Medium Supports the Physiological Activity of Neuronal Tissue in vitro发布日期: 07/22/2016 -
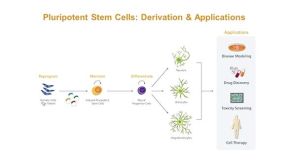
 研究综述Neural Stem Cells: Identification, Function, Culture, and Isolation
研究综述Neural Stem Cells: Identification, Function, Culture, and Isolation细胞类型:
神经干祖细胞
发布日期: 04/01/2015 -
-



 沪公网安备31010102008431号
沪公网安备31010102008431号