
 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




技术资料
-
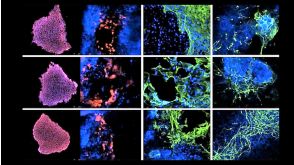
 专家访谈Scott Allen, PhD Seeking Metabolic Therapies for an Incurable Neurodegenerative Disease
专家访谈Scott Allen, PhD Seeking Metabolic Therapies for an Incurable Neurodegenerative Disease研究方向:
干细胞生物学,神经科学
发布日期: 08/29/2018 -
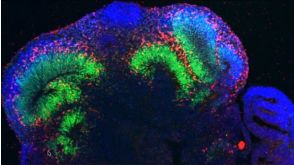
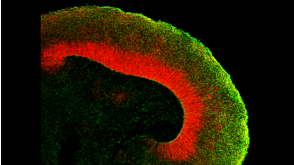
 挂图Building Three-Dimensional Human Brain Organoids Overview of brain organogenesis and the applications of brain organoids in studying the development and maturation of the nervous system发布日期: 05/11/2018
挂图Building Three-Dimensional Human Brain Organoids Overview of brain organogenesis and the applications of brain organoids in studying the development and maturation of the nervous system发布日期: 05/11/2018 -

-
 27:19
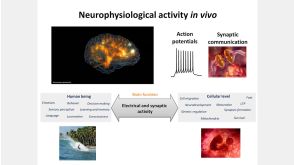
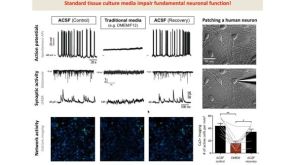
线上讲座BrainPhys™ Medium Supports the Physiological Activity of Neuronal Tissue in vitro发布日期: 07/22/2016
27:19
线上讲座BrainPhys™ Medium Supports the Physiological Activity of Neuronal Tissue in vitro发布日期: 07/22/2016


 沪公网安备31010102008431号
沪公网安备31010102008431号