FUT2-dependent fucosylation of HYOU1 protects intestinal stem cells against inflammatory injury by regulating unfolded protein response.
The intestinal epithelial repair after injury is coordinated by intestinal stem cells (ISCs). Fucosylation catalyzed by fucosyltransferase 2 (FUT2) of the intestinal epithelium is beneficial to mucosal healing but poorly defined is the influence on ISCs. The dextran sulfate sodium (DSS) and lipopolysaccharide (LPS) model were used to assess the role of FUT2 on ISCs after injury. The apoptosis,function,and stemness of ISCs were analyzed using intestinal organoids from WT and Fut2?ISC (ISC-specific Fut2 knockout) mice incubated with LPS and fucose. N-glycoproteomics,UEA-1 chromatography,and site-directed mutagenesis were monitored to dissect the regulatory mechanism,identify the target fucosylated protein and the corresponding modification site. Fucose could alleviate intestinal epithelial damage via upregulating FUT2 and ?-1,2-fucosylation of ISCs. Oxidative stress,mitochondrial dysfunction,and cell apoptosis were impeded by fucose. Meanwhile,fucose sustained the growth and proliferation capacity of intestinal organoids treated with LPS. Contrarily,FUT2 depletion in ISCs aggravated the epithelial damage and disrupted the growth and proliferation capacity of ISCs via escalating LPS-induced endoplasmic reticulum (ER) stress and initiating the IRE1/TRAF2/ASK1/JNK branch of unfolded protein response (UPR). Fucosylation of the chaperone protein HYOU1 at the N-glycosylation site of asparagine (Asn) 862 mediated by FUT2 was identified to facilitate ISCs survival and self-renewal,and improve ISCs resistance to ER stress and inflammatory injury. Our study highlights a fucosylation-dependent protective mechanism of ISCs against inflammation,which may provide a fascinating strategy for treating intestinal injury disorders.
View Publication
D. Jayawardena et al. ( 2023)
Cellular and molecular gastroenterology and hepatology 15 903-919
Loss of SLC26A3 Results in Colonic Mucosal Immune Dysregulation via Epithelial-Immune Cell Crosstalk.
BACKGROUND & AIMS Down-regulation of chloride transporter SLC26A3 or down-regulated in adenoma (DRA) in colonocytes has recently been linked to the pathogenesis of ulcerative colitis (UC). Because exaggerated immune responses are one of the hallmarks of UC,these current studies were undertaken to define the mechanisms by which loss of DRA relays signals to immune cells to increase susceptibility to inflammation. METHODS NanoString Immunology Panel,fluorescence assisted cell sorting,immunoblotting,immunofluorescence,and quantitative real-time polymerase chain reaction assays were used in wild-type and DRA knockout (KO) mice. Interleukin (IL)-33 blocking was used to determine specific changes in immune cells and co-housing/broad spectrum antibiotics administration,and ex vivo studies in colonoids were conducted to rule out the involvement of microbiota. Colonoid-derived monolayers from healthy and UC patient biopsies were analyzed for translatability. RESULTS There was a marked induction of Th2 (>2-fold),CD4+ Th2 cells (~8-fold),RORt+ Th17,and FOXP3+ regulatory T cells (Tregs). DRA KO colons also exhibited a robust induction of IL-33 (>8-fold). In vivo studies using blocking of IL-33 established that T2 immune dysregulation (alterations in ILC2,Th2,and GATA3+ iTregs) in response to loss of DRA was due to altered epithelial-immune cell crosstalk via IL-33. CONCLUSIONS Loss of DRA in colonocytes triggers the release of IL-33 to drive a type 2 immune response. These observations emphasize the critical importance of DRA in mucosal immune homeostasis and its implications in the pathogenesis of UC.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




 专家访谈Tamara Zietek, PhD Studying Intestinal Nutrient Absorption with Organoids
专家访谈Tamara Zietek, PhD Studying Intestinal Nutrient Absorption with Organoids 专家访谈Caroline Lindemans, MD, PhD How Organoids Provide a Model System for Intestinal Regeneration and Repair
专家访谈Caroline Lindemans, MD, PhD How Organoids Provide a Model System for Intestinal Regeneration and Repair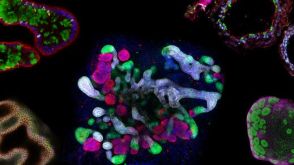 研究综述The Predictive Power of Organoid-Based New Approach Methodologies in Drug Discovery
研究综述The Predictive Power of Organoid-Based New Approach Methodologies in Drug Discovery
 挂图Dynamic Modeling in Organoids Learn more about organoid applications for studying human health
挂图Dynamic Modeling in Organoids Learn more about organoid applications for studying human health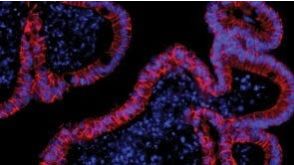




 沪公网安备31010102008431号
沪公网安备31010102008431号