Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells.
During persistent murine cytomegalovirus (MCMV) infection,the T cell response is maintained at extremely high intensity for the life of the host. These cells closely resemble human CMV-specific cells,which compose a major component of the peripheral T cell compartment in most people. Despite a phenotype that suggests extensive antigen-driven differentiation,MCMV-specific T cells remain functional and respond vigorously to viral challenge. We hypothesized that a low rate of antigen-driven proliferation would account for the maintenance of this population. Instead,we found that most of these cells divided only sporadically in chronically infected hosts and had a short half-life in circulation. The overall population was supported,at least in part,by memory T cells primed early in infection,as well as by recruitment of naive T cells at late times. Thus,these data show that memory inflation is maintained by a continuous replacement of short-lived,functional cells during chronic MCMV infection.
View Publication
产品号#:
15023
15063
产品名:
RosetteSep™ 人CD8+ T细胞富集抗体混合物
RosetteSep™人CD8+ T细胞富集抗体混合物
Wu X et al. (DEC 2008)
Blood 112 12 4675--82
Alternative splicing regulates activation-induced cytidine deaminase (AID): implications for suppression of AID mutagenic activity in normal and malignant B cells.
The mutagenic enzyme activation-induced cytidine deaminase (AID) is required for immunoglobulin class switch recombination (CSR) and somatic hypermutation (SHM) in germinal center (GC) B cells. Deregulated expression of AID is associated with various B-cell malignancies and,currently,it remains unclear how AID activity is extinguished to avoid illegitimate mutations. AID has also been shown to be alternatively spliced in malignant B cells,and there is limited evidence that this also occurs in normal blood B cells. The functional significance of these splice variants remains unknown. Here we show that normal GC human B cells and blood memory B cells similarly express AID splice variants and show for the first time that AID splicing variants are singly expressed in individual normal B cells as well as malignant B cells from chronic lymphocytic leukemia patients. We further demonstrate that the alternative AID splice variants display different activities ranging from inactivation of CSR to inactivation or heightened SHM activity. Our data therefore suggest that CSR and SHM are differentially switched off by varying the expression of splicing products of AID at the individual cell level. Most importantly,our findings suggest a novel tumor suppression mechanism by which unnecessary AID mutagenic activities are promptly contained for GC B cells.
View Publication
产品号#:
19054
19054RF
20119
20155
21000
产品名:
EasySep™人B细胞富集试剂盒
RoboSep™ 人B细胞富集试剂盒含滤芯吸头
RoboSep™ 吸头组件抛光剂
RoboSep™分选管套装(9个塑料管)
RoboSep™- S
Fogli M et al. (JUL 2008)
PLoS pathogens 4 7 e1000101
Lysis of endogenously infected CD4+ T cell blasts by rIL-2 activated autologous natural killer cells from HIV-infected viremic individuals.
Understanding the cellular mechanisms that ensure an appropriate innate immune response against viral pathogens is an important challenge of biomedical research. In vitro studies have shown that natural killer (NK) cells purified from healthy donors can kill heterologous cell lines or autologous CD4+ T cell blasts exogenously infected with several strains of HIV-1. However,it is not known whether the deleterious effects of high HIV-1 viremia interferes with the NK cell-mediated cytolysis of autologous,endogenously HIV-1-infected CD4+ T cells. Here,we stimulate primary CD4+ T cells,purified ex vivo from HIV-1-infected viremic patients,with PHA and rIL2 (with or without rIL-7). This experimental procedure allows for the significant expansion and isolation of endogenously infected CD4+ T cell blasts detected by intracellular staining of p24 HIV-1 core antigen. We show that,subsequent to the selective down-modulation of MHC class-I (MHC-I) molecules,HIV-1-infected p24(pos) blasts become partially susceptible to lysis by rIL-2-activated NK cells,while uninfected p24(neg) blasts are spared from killing. This NK cell-mediated killing occurs mainly through the NKG2D activation pathway. However,the degree of NK cell cytolytic activity against autologous,endogenously HIV-1-infected CD4+ T cell blasts that down-modulate HLA-A and -B alleles and against heterologous MHC-I(neg) cell lines is particularly low. This phenomenon is associated with the defective surface expression and engagement of natural cytotoxicity receptors (NCRs) and with the high frequency of the anergic CD56(neg)/CD16(pos) subsets of highly dysfunctional NK cells from HIV-1-infected viremic patients. Collectively,our data demonstrate that the chronic viral replication of HIV-1 in infected individuals results in several phenotypic and functional aberrancies that interfere with the NK cell-mediated killing of autologous p24(pos) blasts derived from primary T cells.
View Publication
产品号#:
19052
19052RF
19055
19055RF
产品名:
EasySep™人CD4+ T细胞富集试剂盒
RoboSep™ 人CD4+ T细胞富集试剂盒含滤芯吸头
EasySep™人NK细胞富集试剂盒
RoboSep™ 人NK细胞富集试剂盒含滤芯吸头
Lambert AA et al. (AUG 2008)
Blood 112 4 1299--307
The C-type lectin surface receptor DCIR acts as a new attachment factor for HIV-1 in dendritic cells and contributes to trans- and cis-infection pathways.
The dynamic interplay between dendritic cells (DCs) and human immunodeficiency virus type-1 (HIV-1) is thought to result in viral dissemination and evasion of antiviral immunity. Although initial observations suggested that the C-type lectin receptor (CLR) DC-SIGN was responsible for the trans-infection function of the virus,subsequent studies demonstrated that trans-infection of CD4(+) T cells with HIV-1 can also occur through DC-SIGN-independent mechanisms. We demonstrate that a cell surface molecule designated DCIR (for DC immunoreceptor),a member of a recently described family of DC-expressing CLRs,can participate in the capture of HIV-1 and promote infection in trans and in cis of autologous CD4(+) T cells from human immature monocyte-derived DCs. The contribution of DCIR to these processes was revealed using DCIR-specific siRNAs and a polyclonal antibody specific for the carbohydrate recognition domain of DCIR. Data from transfection experiments indicated that DCIR acts as a ligand for HIV-1 and is involved in events leading to productive virus infection. Finally,we show that the neck domain of DCIR is important for the DCIR-mediated effect on virus binding and infection. These results point to a possible role for DCIR in HIV-1 pathogenesis by supporting the productive infection of DCs and promoting virus propagation.
View Publication
产品号#:
19052
19052RF
产品名:
EasySep™人CD4+ T细胞富集试剂盒
RoboSep™ 人CD4+ T细胞富集试剂盒含滤芯吸头
Chang SK et al. (JUN 2008)
Journal of immunology (Baltimore,Md. : 1950) 180 11 7394--403
B lymphocyte stimulator regulates adaptive immune responses by directly promoting dendritic cell maturation.
B lymphocyte stimulator (BLyS) is a well-known direct costimulator of adaptive immune cells,particularly B lineage cells. However,we have reported recently that BLyS is also able to activate monocytes. Other innate immune cells,such as dendritic cells (DCs),play a key role in the initiation of adaptive immune responses and the purpose of the current study was to assess whether there is a direct role for BLyS in modulating human DC functions. In this study,we show that BLyS induces DC activation and maturation. Thus,BLyS strongly induced up-regulation of surface costimulatory molecule expression and secretion of specific cytokines and chemokines in DCs. BLyS-stimulated DCs (BLyS-DCs) were also able to augment allogeneic CD4 T cell proliferation to a greater extent than control DCs. BLyS-DCs secreted elevated levels of the major Th1-polarizing cytokine,IL-12p70,and they promoted naive CD4 T cell differentiation into Th1 T cells. Regarding BLyS receptor expression,DCs primarily express cytoplasmic transmembrane activator and CAML interactor; however,low levels of cell surface transmembrane activator and CAML interactor are expressed as well. Collectively,our data suggest that BLyS may modulate adaptive immune cells indirectly by inducing DC maturation.
View Publication
产品号#:
20119
20155
21000
产品名:
RoboSep™ 吸头组件抛光剂
RoboSep™分选管套装(9个塑料管)
RoboSep™- S
Gilbert C et al. (JUL 2007)
Journal of virology 81 14 7672--82
Human immunodeficiency virus type 1 replication in dendritic cell-T-cell cocultures is increased upon incorporation of host LFA-1 due to higher levels of virus production in immature dendritic cells.
Dendritic cells (DCs) act as a portal for invasion by human immunodeficiency virus type-1 (HIV-1). Here,we investigated whether virion-incorporated host cell membrane proteins can affect virus replication in DC-T-cell cocultures. Using isogenic viruses either devoid of or bearing host-derived leukocyte function-associated antigen 1 (LFA-1),we showed that HIV-1 production is augmented when LFA-1-bearing virions are used compared to that for viral entities lacking this adhesion molecule. This phenomenon was observed in immature monocyte-derived DCs (IM-MDDCs) only and not in DCs displaying a mature phenotype. The increase is not due to higher virus production in responder CD4(+) T cells but rather is linked with a more important productive infection of IM-MDDCs. We provided evidence that virus-associated host LFA-1 molecules do not affect a late event in the HIV-1 life cycle but rather exert an effect on an early step in virus replication. We demonstrated that the enhancement of productive infection of IM-MDDCs that is conferred by virus-anchored host LFA-1 involves the protein kinase A (PKA) and PKC signal transduction pathways. The biological significance of this phenomenon was established by performing experiments with virus stocks produced in primary human cells and anti-LFA-1 antibodies. Together,our results indicate that the association between some virus-bound host proteins and their natural cognate ligands can modulate de novo HIV-1 production by IM-MDDCs. Therefore,the additional interactions between virus-bound host cell membrane constituents and counter receptors on the surfaces of DCs can influence HIV-1 replication in IM-MDDC-T-cell cocultures.
View Publication
产品号#:
19052
19052RF
产品名:
EasySep™人CD4+ T细胞富集试剂盒
RoboSep™ 人CD4+ T细胞富集试剂盒含滤芯吸头
Isham CR et al. (MAR 2007)
Blood 109 6 2579--88
Chaetocin: a promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress.
Chaetocin,a thiodioxopiperazine natural product previously unreported to have anticancer effects,was found to have potent antimyeloma activity in IL-6-dependent and -independent myeloma cell lines in freshly collected sorted and unsorted patient CD138(+) myeloma cells and in vivo. Chaetocin largely spares matched normal CD138(-) patient bone marrow leukocytes,normal B cells,and neoplastic B-CLL (chronic lymphocytic leukemia) cells,indicating a high degree of selectivity even in closely lineage-related B cells. Furthermore,chaetocin displays superior ex vivo antimyeloma activity and selectivity than doxorubicin and dexamethasone,and dexamethasone- or doxorubicin-resistant myeloma cell lines are largely non-cross-resistant to chaetocin. Mechanistically,chaetocin is dramatically accumulated in cancer cells via a process inhibited by glutathione and requiring intact/unreduced disulfides for uptake. Once inside the cell,its anticancer activity appears mediated primarily through the imposition of oxidative stress and consequent apoptosis induction. Moreover,the selective antimyeloma effects of chaetocin appear not to reflect differential intracellular accumulation of chaetocin but,instead,heightened sensitivity of myeloma cells to the cytotoxic effects of imposed oxidative stress. Considered collectively,chaetocin appears to represent a promising agent for further study as a potential antimyeloma therapeutic.
View Publication
产品号#:
20119
20155
21000
73592
产品名:
RoboSep™ 吸头组件抛光剂
RoboSep™分选管套装(9个塑料管)
RoboSep™- S
毛壳素
Vieillard V et al. (AUG 2005)
Proceedings of the National Academy of Sciences 102 31 10981--86
NK cytotoxicity against CD4+ T cells during HIV-1 infection: A gp41 peptide induces the expression of an NKp44 ligand
HIV infection leads to a state of chronic immune activation and progressive deterioration in immune function,manifested most recognizably by the progressive depletion of CD4+ T cells. A substantial percentage of natural killer (NK) cells from patients with HIV infection are activated and express the natural cytotoxicity receptor (NCR) NKp44. Here we show that a cellular ligand for NKp44 (NKp44L) is expressed during HIV-1 infection and is correlated with both the progression of CD4+ T cell depletion and the increase of viral load. CD4+ T cells expressing this ligand are highly sensitive to the NK lysis activity mediated by NKp44+ NK cells. The expression of NKp44L is induced by the linear motif NH2-SWSNKS-COOH of the HIV-1 envelope gp41 protein. This highly conserved motif appears critical to the sharp increase in NK lysis of CD4+ T cells from HIV-infected patients. These studies strongly suggest that induction of NKp44L plays a key role in the lysis of CD4+ T cells by activated NK cells in HIV infection and consequently provide a framework for considering how HIV-1 may use NK cell immune surveillance to trigger CD4+ T cells. Understanding this mechanism may help to develop future therapeutic strategies and vaccines against HIV-1 infection.
View Publication
产品号#:
03800
03801
03802
03803
03804
03805
03806
05150
15021
15061
产品名:
ClonaCell™-HY 杂交瘤试剂盒
ClonaCell™-HY Medium
ClonaCell™-HY Medium
ClonaCell™-HY Medium
ClonaCell™-HY Medium
ClonaCell™-HY Medium
ClonaCell™-HY PEG (融合)
MyeloCult™H5100
RosetteSep™人T细胞富集抗体混合物
RosetteSep™人T细胞富集抗体混合物
Feeney ME et al. (DEC 2003)
Journal of immunology (Baltimore,Md. : 1950) 171 12 6968--75
Reconstitution of virus-specific CD4 proliferative responses in pediatric HIV-1 infection.
Gag-specific CD4 proliferative responses correlate inversely with HIV-1 RNA levels in infected adults,and robust responses are characteristic of long-term nonprogressive infection. However,strong responses are seldom detected in adult subjects with progressive infection and are not generally reconstituted on highly active antiretroviral therapy (HAART). To date,the role of HIV-1-specific Th responses in children has not been thoroughly examined. We characterized Gag-specific CD4 responses among 35 perinatally infected subjects,including 2 children who spontaneously control viremia without antiretroviral therapy,21 children with viral loads (VL) of textless400 on HAART,and 12 viremic children. Gag-specific Th activity was assessed by lymphoproliferative assay,and responses were mapped using overlapping Gag peptides in an IFN-gamma ELISPOT. Robust proliferative responses were detected in the children exhibiting spontaneous control of viremia,and mapping of targeted Gag regions in one such subject identified multiple epitopes. Among children textgreateror=5 years old,14 of 17 subjects with VL of textless400 on HAART demonstrated a significant p24 proliferative response (median p24 stimulation index,20),in contrast with only 1 of 9 viremic children (median p24 stimulation index,2.0; p = 0.0008). However,no subject younger than 5 years of age possessed a significant response,even when viremia was fully suppressed. When compared with adults with VL of textless400 on HAART,Th responses among children with VL of textless400 were both more frequent (p = 0.009) and of greater magnitude (p = 0.002). These data suggest that children may have a greater intrinsic capacity to reconstitute HIV-1-specific immunity than adults,and may be excellent candidates for immune-based therapies.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




 挂图The Immune Response to HIV Poster Summary of how HIV subverts the immune response to establish a chronic infection
挂图The Immune Response to HIV Poster Summary of how HIV subverts the immune response to establish a chronic infection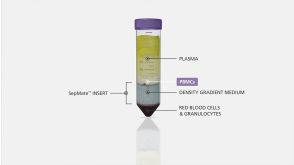

 沪公网安备31010102008431号
沪公网安备31010102008431号