Woods CM et al. ( 1995)
Molecular medicine (Cambridge,Mass.) 1 5 506--526
Taxol-induced mitotic block triggers rapid onset of a p53-independent apoptotic pathway.
BACKGROUND: At therapeutic concentrations,the antineoplastic agent taxol selectively perturbs mitotic spindle microtubules. Taxol has recently been shown to induce apoptosis,similar to the mechanism of cell death induced by other antineoplastic agents. However,taxol has shown efficacy against drug-refractory cancers,raising the possibility that this pharmacological agent may trigger an alternative apoptotic pathway. MATERIALS AND METHODS: The kinetics and IC50 of mitotic (M) block,aberrant mitosis,and cytotoxicity following taxol treatment were analyzed in human cell lines as well as normal mouse embryo fibroblasts (MEFs) and MEFs derived from p53-null mice. Apoptosis was followed by DNA gel electrophoresis and by in situ DNA end-labeling (TUNEL). RESULTS: Taxol induced two forms of cell cycle arrest: either directly in early M at prophase or,for those cells progressing through aberrant mitosis,arrest in G1 as multimininucleated cells. TUNEL labeling revealed that DNA nicking occurred within 30 min of the arrest in prophase. In contrast,G1-arrested,multimininucleated cells became TUNEL positive only after several days. In the subset of cells that became blocked directly in prophase,both wt p53-expressing and p53-null MEFs responded similarly to taxol,showing rapid onset of DNA nicking and apoptosis. However,p53-null MEFs progressing through aberrant mitosis failed to arrest in the subsequent G1 phase or to become TUNEL positive,and remained viable. CONCLUSIONS: Taxol induces two forms of cell cycle arrest,which in turn induce two independent apoptotic pathways. Arrest in prophase induces rapid onset of a p53-independent pathway,whereas G1-block and the resulting slow (3-5 days) apoptotic pathway are p53 dependent.
View Publication
产品号#:
73312
73314
产品名:
紫杉醇
紫杉醇
Blagosklonny MV et al. ( 1995)
Cancer research 55 20 4623--4626
Taxol induction of p21WAF1 and p53 requires c-raf-1.
Taxol stabilizes microtubules,prevents tubulin depolymerization,and promotes tubulin bundling and is one of the most effective drugs for the treatment of metastatic breast and ovarian cancer. Although its interaction with tubulin has been well characterized,the mechanism by which taxol induces growth arrest and cytotoxicity is not well understood. Herein,we show that taxol induced dose- and time-dependent accumulation of the cyclin inhibitor p21WAF1 in both p53 wild-type and p53-null cells,although the degree of induction was greater in cells expressing wild-type p53. In MCF7 cells,wild-type p53 protein was also induced after taxol treatment,and this induction was mediated primarily by increased protein stability. Taxol induced both p21WAF1 and wild-type p53 optimally in MCF7 cells after 20-24-h exposure with an EC50(3) of 5 nM. In p53-null PC3M cells,p21WAF1 was similarly induced after 24-h exposure to taxol. Coincident with these biochemical effects,taxol altered the electrophoretic mobility of c-raf-1 and stimulated mitogen activated protein kinase. Previous depletion of c-raf-1 inhibited both the p21WAF1- and p53-inducing properties of taxol,as well as the activation of MAP kinase. These data suggest that induction of p21WAF1 by taxol requires c-raf-1 activity,but that it is not strictly dependent on wild-type p53. Furthermore,the ability of taxol to both induce wild-type p53 in MCF7 cells and activate MAP kinase is also dependent on c-raf-1 expression.
View Publication
产品号#:
73312
73314
产品名:
紫杉醇
紫杉醇
Kanzaki H et al. ( 2016)
Scientific Reports 6 August 32259
A-Disintegrin and Metalloproteinase (ADAM) 17 enzymatically degrades interferon-gamma
Interferon-gamma (IFN-γ) is a pleiotropic cytokine that exerts anti-tumor and anti-osteoclastogenic effects. Although transcriptional and post-transcriptional regulation of IFN-γ is well understood,subsequent modifications of secreted IFN-γ are not fully elucidated. Previous research indicates that some cancer cells escape immune surveillance and metastasize into bone tissue by inducing osteoclastic bone resorption. Peptidases of the a-disintegrin and metalloproteinase (ADAM) family are implicated in cancer cell proliferation and tumor progression. We hypothesized that the ADAM enzymes expressed by cancer cells degrades IFN-γ and attenuates IFN-γ-mediated anti-tumorigenic and anti-osteoclastogenic effects. Recombinant ADAM17 degraded IFN-γ into small fragments. The addition of ADAM17 to the culture supernatant of stimulated mouse splenocytes decreased IFN-γ concentration. However,ADAM17 inhibition in the stimulated mouse T-cells prevented IFN-γ degradation. ADAM17-expressing human breast cancer cell lines MCF-7 and MDA-MB-453 also degraded recombinant IFN-γ,but this was attenuated by ADAM17 inhibition. Degraded IFN-γ lost the functionality including the inhibititory effect on osteoclastogenesis. This is the first study to demonstrate the extracellular proteolytic degradation of IFN-γ by ADAM17. These results suggest that ADAM17-mediated degradation of IFN-γ may block the anti-tumorigenic and anti-osteoclastogenic effects of IFN-γ. ADAM17 inhibition may be useful for the treatment of attenuated cancer immune surveillance and/or bone metastases.
View Publication
产品号#:
03800
产品名:
ClonaCell™-HY杂交瘤试剂盒
Li T et al. ( 2016)
Scientific reports 6 27055
Immuno-targeting the multifunctional CD38 using nanobody.
CD38,as a cell surface antigen is highly expressed in several hematologic malignancies including multiple myeloma (MM) and has been proven to be a good target for immunotherapy of the disease. CD38 is also a signaling enzyme responsible for the metabolism of two novel calcium messenger molecules. To be able to target this multifunctional protein,we generated a series of nanobodies against CD38 with high affinities. Crystal structures of the complexes of CD38 with the nanobodies were solved,identifying three separate epitopes on the carboxyl domain. Chromobodies,engineered by tagging the nanobody with fluorescence proteins,provide fast,simple and versatile tools for quantifying CD38 expression. Results confirmed that CD38 was highly expressed in malignant MM cells compared with normal white blood cells. The immunotoxin constructed by splicing the nanobody with a bacterial toxin,PE38 shows highly selective cytotoxicity against patient-derived MM cells as well as the cell lines,with half maximal effective concentration reaching as low as 10(-11) molar. The effectiveness of the immunotoxin can be further increased by stimulating CD38 expression using retinoid acid. These results set the stage for the development of clinical therapeutics as well as diagnostic screening for myeloma.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒




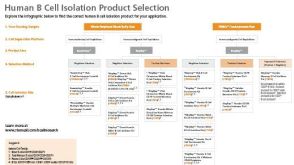
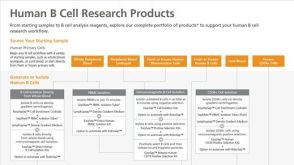



 科学海报Rapid Expansion of Functional Human T Cells Using a Novel Serum-Free and Xeno-Free Culture Medium
科学海报Rapid Expansion of Functional Human T Cells Using a Novel Serum-Free and Xeno-Free Culture Medium


 沪公网安备31010102008431号
沪公网安备31010102008431号