Wattanapanitch M et al. (SEP 2014)
PloS one 9 9 e106952
Dual small-molecule targeting of SMAD signaling stimulates human induced pluripotent stem cells toward neural lineages.
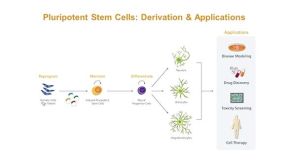
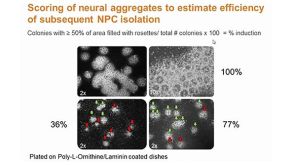
Incurable neurological disorders such as Parkinson's disease (PD),Huntington's disease (HD),and Alzheimer's disease (AD) are very common and can be life-threatening because of their progressive disease symptoms with limited treatment options. To provide an alternative renewable cell source for cell-based transplantation and as study models for neurological diseases,we generated induced pluripotent stem cells (iPSCs) from human dermal fibroblasts (HDFs) and then differentiated them into neural progenitor cells (NPCs) and mature neurons by dual SMAD signaling inhibitors. Reprogramming efficiency was improved by supplementing the histone deacethylase inhibitor,valproic acid (VPA),and inhibitor of p160-Rho associated coiled-coil kinase (ROCK),Y-27632,after retroviral transduction. We obtained a number of iPS colonies that shared similar characteristics with human embryonic stem cells in terms of their morphology,cell surface antigens,pluripotency-associated gene and protein expressions as well as their in vitro and in vivo differentiation potentials. After treatment with Noggin and SB431542,inhibitors of the SMAD signaling pathway,HDF-iPSCs demonstrated rapid and efficient differentiation into neural lineages. Six days after neural induction,neuroepithelial cells (NEPCs) were observed in the adherent monolayer culture,which had the ability to differentiate further into NPCs and neurons,as characterized by their morphology and the expression of neuron-specific transcripts and proteins. We propose that our study may be applied to generate neurological disease patient-specific iPSCs allowing better understanding of disease pathogenesis and drug sensitivity assays.
View Publication
产品号#:
05850
05857
05870
05875
07923
85850
85857
85870
85875
产品名:
Dispase (1 U/mL)
mTeSR™1
mTeSR™1
Zhou J et al. (AUG 2016)
Neurochemical Research 41 8 2065--2074
Generation of Human Embryonic Stem Cell Line Expressing zsGreen in Cholinergic Neurons Using CRISPR/Cas9 System
Lineage specific human embryonic stem cell (hESC) reporter cell line is a versatile tool for biological studies on real time monitoring of differentiation,physiological and biochemical features of special cell types and pathological mechanism of disease. Here we report the generation of ChAT-zsGreen reporter hESC line that express zsGreen under the control of the choline acetyltransferase (ChAT) promoter using CRISPR (Clustered Regularly Interspersed Short Palindromic Repeats)/Cas9 system. We show that the ChAT-zsGreen hESC reporter cell lines retain the features of undifferentiated hESC. After cholinergic neuronal differentiation,cholinergic neurons were clearly labeled with green fluorescence protein (zsGreen). The ChAT-zsGreen reporter hESC lines are invaluable not only for the monitoring cholinergic neuronal differentiation but also for study physiological and biochemical hallmarks of cholinergic neurons.
View Publication
产品号#:
05940
产品名:
Busskamp V et al. (NOV 2014)
Molecular systems biology 10 11 760
Rapid neurogenesis through transcriptional activation in human stem cells.
Advances in cellular reprogramming and stem cell differentiation now enable ex vivo studies of human neuronal differentiation. However,it remains challenging to elucidate the underlying regulatory programs because differentiation protocols are laborious and often result in low neuron yields. Here,we overexpressed two Neurogenin transcription factors in human-induced pluripotent stem cells and obtained neurons with bipolar morphology in 4 days,at greater than 90% purity. The high purity enabled mRNA and microRNA expression profiling during neurogenesis,thus revealing the genetic programs involved in the rapid transition from stem cell to neuron. The resulting cells exhibited transcriptional,morphological and functional signatures of differentiated neurons,with greatest transcriptional similarity to prenatal human brain samples. Our analysis revealed a network of key transcription factors and microRNAs that promoted loss of pluripotency and rapid neurogenesis via progenitor states. Perturbations of key transcription factors affected homogeneity and phenotypic properties of the resulting neurons,suggesting that a systems-level view of the molecular biology of differentiation may guide subsequent manipulation of human stem cells to rapidly obtain diverse neuronal types.
View Publication
产品号#:
05854
05855
05850
05857
05870
05875
85850
85857
85870
85875
产品名:
mFreSR™
mFreSR™
mTeSR™1
mTeSR™1
Devlin A-C et al. (JAN 2015)
Nature Communications 6 1--12
Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability
Crook JM et al. (MAR 2015)
Expert review of neurotherapeutics 15 3 295--304
The potential of induced pluripotent stem cells in models of neurological disorders: implications on future therapy.
There is an urgent need for new and advanced approaches to modeling the pathological mechanisms of complex human neurological disorders. This is underscored by the decline in pharmaceutical research and development efficiency resulting in a relative decrease in new drug launches in the last several decades. Induced pluripotent stem cells represent a new tool to overcome many of the shortcomings of conventional methods,enabling live human neural cell modeling of complex conditions relating to aberrant neurodevelopment,such as schizophrenia,epilepsy and autism as well as age-associated neurodegeneration. This review considers the current status of induced pluripotent stem cell-based modeling of neurological disorders,canvassing proven and putative advantages,current constraints,and future prospects of next-generation culture systems for biomedical research and translation.
View Publication
产品号#:
05850
05857
05870
05875
85850
85857
85870
85875
产品名:
mTeSR™1
mTeSR™1
Takayama Y and Kida YS (FEB 2016)
PloS one 11 2 e0148559
In Vitro Reconstruction of Neuronal Networks Derived from Human iPS Cells Using Microfabricated Devices.
Morphology and function of the nervous system is maintained via well-coordinated processes both in central and peripheral nervous tissues,which govern the homeostasis of organs/tissues. Impairments of the nervous system induce neuronal disorders such as peripheral neuropathy or cardiac arrhythmia. Although further investigation is warranted to reveal the molecular mechanisms of progression in such diseases,appropriate model systems mimicking the patient-specific communication between neurons and organs are not established yet. In this study,we reconstructed the neuronal network in vitro either between neurons of the human induced pluripotent stem (iPS) cell derived peripheral nervous system (PNS) and central nervous system (CNS),or between PNS neurons and cardiac cells in a morphologically and functionally compartmentalized manner. Networks were constructed in photolithographically microfabricated devices with two culture compartments connected by 20 microtunnels. We confirmed that PNS and CNS neurons connected via synapses and formed a network. Additionally,calcium-imaging experiments showed that the bundles originating from the PNS neurons were functionally active and responded reproducibly to external stimuli. Next,we confirmed that CNS neurons showed an increase in calcium activity during electrical stimulation of networked bundles from PNS neurons in order to demonstrate the formation of functional cell-cell interactions. We also confirmed the formation of synapses between PNS neurons and mature cardiac cells. These results indicate that compartmentalized culture devices are promising tools for reconstructing network-wide connections between PNS neurons and various organs,and might help to understand patient-specific molecular and functional mechanisms under normal and pathological conditions.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒







 沪公网安备31010102008431号
沪公网安备31010102008431号