Sulforaphane targets pancreatic tumour-initiating cells by NF-kappaB-induced antiapoptotic signalling.
BACKGROUND AND AIMS: Emerging evidence suggests that highly treatment-resistant tumour-initiating cells (TICs) play a central role in the pathogenesis of pancreatic cancer. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) is considered to be a novel anticancer agent; however,recent studies have shown that many pancreatic cancer cells are resistant to apoptosis induction by TRAIL due to TRAIL-activated nuclear factor-kappaB (NF-kappaB) signalling. Several chemopreventive agents are able to inhibit NF-kappaB,and favourable results have been obtained--for example,for the broccoli compound sulforaphane-in preventing metastasis in clinical studies. The aim of the study was to identify TICs in pancreatic carcinoma for analysis of resistance mechanisms and for definition of sensitising agents. METHODS: TICs were defined by expression patterns of a CD44(+)/CD24(-),CD44(+)/CD24(+) or CD44(+)/CD133(+) phenotype and correlation to growth in immunodeficient mice,differentiation grade,clonogenic growth,sphere formation,aldehyde dehydrogenase (ALDH) activity and therapy resistance. RESULTS: Mechanistically,specific binding of transcriptionally active cRel-containing NF-kappaB complexes in TICs was observed. Sulforaphane prevented NF-kappaB binding,downregulated apoptosis inhibitors and induced apoptosis,together with prevention of clonogenicity. Gemcitabine,the chemopreventive agents resveratrol and wogonin,and the death ligand TRAIL were less effective. In a xenograft model,sulforaphane strongly blocked tumour growth and angiogenesis,while combination with TRAIL had an additive effect without obvious cytotoxicity in normal cells. Freshly isolated patient tumour cells expressing markers for TICs could be sensitised by sulforaphane for TRAIL-induced cytotoxicity. CONCLUSION: The data provide new insights into resistance mechanisms of TICs and suggest the combination of sulforaphane with TRAIL as a promising strategy for targeting of pancreatic TICs.
View Publication
Lee Y-KK et al. (JAN 2016)
International journal of cardiology 203 964--971
Efficient attenuation of Friedreich's ataxia (FRDA) cardiomyopathy by modulation of iron homeostasis-human induced pluripotent stem cell (hiPSC) as a drug screening platform for FRDA.
BACKGROUND Friedreich's ataxia (FRDA),a recessive neurodegenerative disorder commonly associated with hypertrophic cardiomyopathy,is caused by silencing of the frataxin (FXN) gene encoding the mitochondrial protein involved in iron-sulfur cluster biosynthesis. METHODS Application of our previously established FRDA human induced pluripotent stem cell (hiPSC) derived cardiomyocytes model as a platform to assess the efficacy of treatment with either the antioxidant coenzyme Q10 analog,idebenone (IDE) or the iron chelator,deferiprone (DFP),which are both under clinical trial. RESULTS DFP was able to more significantly suppress synthesis of reactive oxygen species (ROS) than IDE at the dosages of 25 $\$ and 10nM respectively which agreed with the reduced rate of intracellular accumulation of iron by DFP treatment from 25 to 50 $\$ With regard to cardiac electrical-contraction (EC) coupling function,decay velocity of calcium handling kinetics in FRDA-hiPSC-cardiomyocytes was significantly improved by DFP treatment but not by IDE. Further mechanistic studies revealed that DFP also modulated iron induced mitochondrial stress as reflected by mitochondria network disorganization and decline level of respiratory chain protein,succinate dehydrogenase (CxII) and cytochrome c oxidase (COXIV). In addition,iron-response protein (IRP-1) regulatory loop was overridden by DFP as reflected by resumed level of ferritin (FTH) back to basal level and the attenuated transferrin receptor (TSFR) mRNA level suppression thereby reducing further iron uptake. CONCLUSIONS DFP modulated iron homeostasis in FRDA-hiPSC-cardiomyocytes and effectively relieved stress-stimulation related to cardiomyopathy. The resuming of redox condition led to the significantly improved cardiac prime events,cardiac electrical-coupling during contraction.
View Publication
Chen C et al. (JUL 2014)
Nature communications 5 4430
Role of astroglia in Down's syndrome revealed by patient-derived human-induced pluripotent stem cells.
Down's syndrome (DS),caused by trisomy of human chromosome 21,is the most common genetic cause of intellectual disability. Here we use induced pluripotent stem cells (iPSCs) derived from DS patients to identify a role for astrocytes in DS pathogenesis. DS astroglia exhibit higher levels of reactive oxygen species and lower levels of synaptogenic molecules. Astrocyte-conditioned medium collected from DS astroglia causes toxicity to neurons,and fails to promote neuronal ion channel maturation and synapse formation. Transplantation studies show that DS astroglia do not promote neurogenesis of endogenous neural stem cells in vivo. We also observed abnormal gene expression profiles from DS astroglia. Finally,we show that the FDA-approved antibiotic drug,minocycline,partially corrects the pathological phenotypes of DS astroglia by specifically modulating the expression of S100B,GFAP,inducible nitric oxide synthase,and thrombospondins 1 and 2 in DS astroglia. Our studies shed light on the pathogenesis and possible treatment of DS by targeting astrocytes with a clinically available drug.
View Publication
Small molecule XIAP inhibitors cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells and overcome Bcl-2-mediated resistance.
Defects in apoptosis contribute to poor outcome in pediatric acute lymphoblastic leukemia (ALL),calling for novel strategies that counter apoptosis resistance. Here,we demonstrate for the first time that small molecule inhibitors of the antiapoptotic protein XIAP cooperate with TRAIL to induce apoptosis in childhood acute leukemia cells. XIAP inhibitors at subtoxic concentrations,but not a structurally related control compound,synergize with TRAIL to trigger apoptosis and to inhibit clonogenic survival of acute leukemia cells,whereas they do not affect viability of normal peripheral blood lymphocytes,suggesting some tumor selectivity. Analysis of signaling pathways reveals that XIAP inhibitors enhance TRAIL-induced activation of caspases,loss of mitochondrial membrane potential,and cytochrome c release in a caspase-dependent manner,indicating that they promote a caspase-dependent feedback mitochondrial amplification loop. Of note,XIAP inhibitors even overcome Bcl-2-mediated resistance to TRAIL by enhancing Bcl-2 cleavage and Bak conformational change. Importantly,XIAP inhibitors kill leukemic blasts from children with ALL ex vivo and cooperate with TRAIL to induce apoptosis. In vivo,they significantly reduce leukemic burden in a mouse model of pediatric ALL engrafted in non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice. Thus,XIAP inhibitors present a promising novel approach for apoptosis-based therapy of childhood ALL.
View Publication
产品类型:
产品号#:
04100
产品名:
MethoCult™ H4100
Puissant A et al. (FEB 2010)
Cancer research 70 3 1042--52
Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation.
Autophagy that is induced by starvation or cellular stress can enable cancer cell survival by sustaining energy homeostasis and eliminating damaged organelles and proteins. In response to stress,cancer cells have been reported to accumulate the protein p62/SQSTM1 (p62),but its role in the regulation of autophagy is controversial. Here,we report that the plant phytoalexin resveratrol (RSV) triggers autophagy in imatinib-sensitive and imatinib-resistant chronic myelogenous leukemia (CML) cells via JNK-dependent accumulation of p62. JNK inhibition or p62 knockdown prevented RSV-mediated autophagy and antileukemic effects. RSV also stimulated AMPK,thereby inhibiting the mTOR pathway. AMPK knockdown or mTOR overexpression impaired RSV-induced autophagy but not JNK activation. Lastly,p62 expression and autophagy in CD34+ progenitors from patients with CML was induced by RSV,and disrupting autophagy protected CD34+ CML cells from RSV-mediated cell death. We concluded that RSV triggered autophagic cell death in CML cells via both JNK-mediated p62 overexpression and AMPK activation. Our findings show that the JNK and AMPK pathways can cooperate to eliminate CML cells via autophagy.
View Publication

 EasySep™小鼠TIL(CD45)正选试剂盒
EasySep™小鼠TIL(CD45)正选试剂盒





 产品手册AggreWell™ for Spheroids
产品手册AggreWell™ for Spheroids 科学海报A Feeder-Free and Serum-Free Culture System Supports Expansion of CD34+ AML and CML Stem/Progenitor Cells
科学海报A Feeder-Free and Serum-Free Culture System Supports Expansion of CD34+ AML and CML Stem/Progenitor Cells 科学海报Manipulation of the Standard CFU-GM Assay Targeted Screening of Hematopoietic Myeloid Progenitors
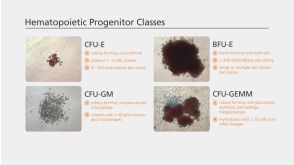
科学海报Manipulation of the Standard CFU-GM Assay Targeted Screening of Hematopoietic Myeloid Progenitors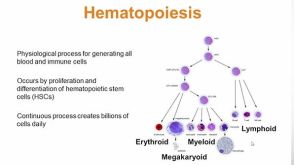



 沪公网安备31010102008431号
沪公网安备31010102008431号